Biopython - Module BioSQL
BioSQLest un schéma de base de données générique conçu principalement pour stocker des séquences et ses données associées pour tous les moteurs SGBDR. Il est conçu de manière à contenir les données de toutes les bases de données bioinformatiques populaires telles que GenBank, Swissport, etc. Il peut également être utilisé pour stocker des données internes.
BioSQL fournit actuellement un schéma spécifique pour les bases de données ci-dessous -
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Il fournit également un support minimal pour les bases de données Java HSQLDB et Derby.
BioPython fournit des capacités ORM très simples, faciles et avancées pour travailler avec une base de données basée sur BioSQL. BioPython provides a module, BioSQL pour faire la fonctionnalité suivante -
- Créer / supprimer une base de données BioSQL
- Connectez-vous à une base de données BioSQL
- Analysez une base de données de séquences comme GenBank, Swisport, résultat BLAST, résultat Entrez, etc., et chargez-la directement dans la base de données BioSQL
- Récupérez les données de séquence de la base de données BioSQL
- Récupérez les données de taxonomie de NCBI BLAST et stockez-les dans la base de données BioSQL
- Exécutez n'importe quelle requête SQL sur la base de données BioSQL
Présentation du schéma de base de données BioSQL
Avant d'approfondir le BioSQL, laissez-nous comprendre les bases du schéma BioSQL. Le schéma BioSQL fournit plus de 25 tables pour contenir des données de séquence, une fonction de séquence, une catégorie de séquence / ontologie et des informations de taxonomie. Certains des tableaux importants sont les suivants -
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
Créer une base de données BioSQL
Dans cette section, créons un exemple de base de données BioSQL, biosql en utilisant le schéma fourni par l'équipe BioSQL. Nous travaillerons avec la base de données SQLite car il est vraiment facile de démarrer et n'a pas de configuration complexe.
Ici, nous allons créer une base de données BioSQL basée sur SQLite en utilisant les étapes ci-dessous.
Step 1 - Téléchargez le moteur de base de données SQLite et installez-le.
Step 2 - Téléchargez le projet BioSQL à partir de l'URL GitHub. https://github.com/biosql/biosql
Step 3 - Ouvrez une console et créez un répertoire en utilisant mkdir et entrez-y.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 - Exécutez la commande ci-dessous pour créer une nouvelle base de données SQLite.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 - Copiez le fichier biosqldb-sqlite.sql du projet BioSQL (/ sql / biosqldb-sqlite.sql`) et stockez-le dans le répertoire courant.
Step 6 - Exécutez la commande ci-dessous pour créer toutes les tables.
sqlite> .read biosqldb-sqlite.sqlDésormais, toutes les tables sont créées dans notre nouvelle base de données.
Step 7 - Exécutez la commande ci-dessous pour voir toutes les nouvelles tables de notre base de données.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>Les trois premières commandes sont des commandes de configuration pour configurer SQLite pour afficher le résultat de manière formatée.
Step 8 - Copiez le fichier d'exemple GenBank, ls_orchid.gbk fourni par l'équipe BioPython https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk dans le répertoire courant et enregistrez-le sous orchid.gbk.
Step 9 - Créez un script python, load_orchid.py en utilisant le code ci-dessous et exécutez-le.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Le code ci-dessus analyse l'enregistrement dans le fichier et le convertit en objets python et l'insère dans la base de données BioSQL. Nous analyserons le code dans la section suivante.
Enfin, nous avons créé une nouvelle base de données BioSQL et y avons chargé des exemples de données. Nous discuterons des tableaux importants dans le chapitre suivant.
Diagramme ER simple
biodatabase table se trouve en haut de la hiérarchie et son objectif principal est d'organiser un ensemble de données de séquence en un seul groupe / base de données virtuelle. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. Toutes les tables associées dans la base de données BioSQL ont des références à l'entrée de la biodatabase.
bioentrytable contient tous les détails d'une séquence à l'exception des données de séquence. données de séquence d'un particulierbioentry sera stocké dans biosequence table.
taxon et taxon_name sont des détails de taxonomie et chaque entrée fait référence à cette table pour spécifier ses informations de taxon.
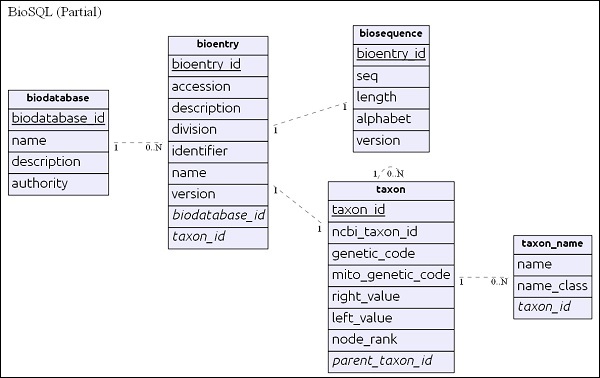
Après avoir compris le schéma, examinons quelques requêtes dans la section suivante.
Requêtes BioSQL
Examinons quelques requêtes SQL pour mieux comprendre comment les données sont organisées et les tables sont liées les unes aux autres. Avant de continuer, ouvrons la base de données en utilisant la commande ci-dessous et définissons quelques commandes de formatage -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Vous pouvez également utiliser n'importe quel éditeur SQLite pour exécuter la requête.
Répertoriez la base de données de séquences virtuelles disponible dans le système comme indiqué ci-dessous -
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Ici, nous n'avons qu'une seule base de données, orchid.
Liste des entrées (3 premiers) disponibles dans la base de données orchid avec le code ci-dessous
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Énumérez les détails de séquence associés à une entrée (accession - Z78530, nom - gène de l'ARNr C. fasciculatum 5.8S et ADN ITS1 et ITS2) avec le code donné -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Obtenez la séquence complète associée à une entrée (accession - Z78530, nom - gène d'ARNr C. fasciculatum 5.8S et ADN ITS1 et ITS2) en utilisant le code ci-dessous -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Liste des taxons associés à la base de données bio, orchidée
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>Charger des données dans la base de données BioSQL
Apprenons à charger des données de séquence dans la base de données BioSQL dans ce chapitre. Nous avons déjà le code pour charger des données dans la base de données dans la section précédente et le code est le suivant -
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Nous examinerons plus en détail chaque ligne du code et son objectif -
Line 1 - Charge le module SeqIO.
Line 2- Charge le module BioSeqDatabase. Ce module fournit toutes les fonctionnalités pour interagir avec la base de données BioSQL.
Line 3 - Charge le module os.
Line 5- open_database ouvre la base de données spécifiée (db) avec le pilote configuré (pilote) et renvoie un handle vers la base de données BioSQL (serveur). Biopython prend en charge les bases de données sqlite, mysql, postgresql et oracle.
Line 6-10- La méthode load_database_sql charge le sql du fichier externe et l'exécute. La méthode commit valide la transaction. Nous pouvons ignorer cette étape car nous avons déjà créé la base de données avec le schéma.
Line 12 - Les méthodes new_database créent une nouvelle base de données virtuelle, orchid et renvoie un handle db pour exécuter la commande sur la base de données orchid.
Line 13- La méthode load charge les entrées de séquence (itérable SeqRecord) dans la base de données orchidée. SqlIO.parse analyse la base de données GenBank et renvoie toutes les séquences qu'elle contient en tant que SeqRecord itérable. Le deuxième paramètre (True) de la méthode de chargement lui demande de récupérer les détails de taxonomie des données de séquence à partir du site Web NCBI Blast, s'il n'est pas déjà disponible dans le système.
Line 14 - commit valide la transaction.
Line 15 - close ferme la connexion à la base de données et détruit le descripteur du serveur.
Récupérer les données de séquence
Récupérons une séquence avec l'identifiant, 2765658 de la base de données d'orchidées comme ci-dessous -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Ici, le serveur ["orchid"] renvoie le handle pour récupérer les données de la base de données virtuelle ouchid. lookup La méthode fournit une option pour sélectionner des séquences en fonction de critères et nous avons sélectionné la séquence avec l'identificateur, 2765658. lookuprenvoie les informations de séquence sous la forme SeqRecordobject. Depuis, nous savons déjà comment travailler avec SeqRecord`, il est facile d'en obtenir des données.
Supprimer une base de données
La suppression d'une base de données est aussi simple que d'appeler la méthode remove_database avec le nom de base de données approprié, puis de la valider comme spécifié ci-dessous -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()