Biopython - Mô-đun BioSQL
BioSQLlà một lược đồ cơ sở dữ liệu chung được thiết kế chủ yếu để lưu trữ trình tự và dữ liệu liên quan của nó cho tất cả công cụ RDBMS. Nó được thiết kế theo cách giữ dữ liệu từ tất cả các cơ sở dữ liệu tin sinh học phổ biến như GenBank, Swissport, v.v. Nó cũng có thể được sử dụng để lưu trữ dữ liệu nội bộ.
BioSQL hiện đang cung cấp lược đồ cụ thể cho các cơ sở dữ liệu bên dưới -
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Nó cũng cung cấp hỗ trợ tối thiểu cho cơ sở dữ liệu HSQLDB và Derby dựa trên Java.
BioPython cung cấp khả năng ORM rất đơn giản, dễ dàng và nâng cao để làm việc với cơ sở dữ liệu dựa trên BioSQL. BioPython provides a module, BioSQL để thực hiện chức năng sau -
- Tạo / xóa cơ sở dữ liệu BioSQL
- Kết nối với cơ sở dữ liệu BioSQL
- Phân tích cú pháp cơ sở dữ liệu trình tự như GenBank, Swisport, kết quả BLAST, kết quả Entrez, v.v. và tải trực tiếp nó vào cơ sở dữ liệu BioSQL
- Tìm nạp dữ liệu trình tự từ cơ sở dữ liệu BioSQL
- Tìm nạp dữ liệu phân loại từ NCBI BLAST và lưu trữ trong cơ sở dữ liệu BioSQL
- Chạy bất kỳ truy vấn SQL nào đối với cơ sở dữ liệu BioSQL
Tổng quan về lược đồ cơ sở dữ liệu BioSQL
Trước khi đi sâu vào BioSQL, chúng ta hãy hiểu cơ bản về lược đồ BioSQL. Lược đồ BioSQL cung cấp hơn 25 bảng để chứa dữ liệu trình tự, tính năng trình tự, danh mục trình tự / bản thể học và thông tin phân loại. Một số bảng quan trọng như sau:
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
Tạo cơ sở dữ liệu BioSQL
Trong phần này, chúng ta hãy tạo một cơ sở dữ liệu BioSQL mẫu, biosql bằng cách sử dụng lược đồ do nhóm BioSQL cung cấp. Chúng tôi sẽ làm việc với cơ sở dữ liệu SQLite vì nó thực sự dễ bắt đầu và không cần thiết lập phức tạp.
Ở đây, chúng tôi sẽ tạo cơ sở dữ liệu BioSQL dựa trên SQLite bằng cách sử dụng các bước dưới đây.
Step 1 - Tải xuống công cụ dữ liệu SQLite và cài đặt nó.
Step 2 - Tải xuống dự án BioSQL từ URL GitHub. https://github.com/biosql/biosql
Step 3 - Mở bảng điều khiển và tạo một thư mục bằng mkdir và nhập vào đó.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 - Chạy lệnh dưới đây để tạo cơ sở dữ liệu SQLite mới.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 - Sao chép tệp biosqldb-sqlite.sql từ dự án BioSQL (/ sql / biosqldb-sqlite.sql`) và lưu trữ trong thư mục hiện tại.
Step 6 - Chạy lệnh dưới đây để tạo tất cả các bảng.
sqlite> .read biosqldb-sqlite.sqlBây giờ, tất cả các bảng được tạo trong cơ sở dữ liệu mới của chúng tôi.
Step 7 - Chạy lệnh dưới đây để xem tất cả các bảng mới trong cơ sở dữ liệu của chúng tôi.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>Ba lệnh đầu tiên là các lệnh cấu hình để cấu hình SQLite hiển thị kết quả theo cách đã định dạng.
Step 8 - Sao chép tệp GenBank mẫu, ls_orchid.gbk do nhóm BioPython cung cấp https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk vào thư mục hiện tại và lưu nó dưới dạng lan.gbk.
Step 9 - Tạo một tập lệnh python, load_orchid.py bằng cách sử dụng mã bên dưới và thực thi nó.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Đoạn mã trên phân tích cú pháp bản ghi trong tệp và chuyển đổi nó thành các đối tượng python và chèn nó vào cơ sở dữ liệu BioSQL. Chúng tôi sẽ phân tích mã trong phần sau.
Cuối cùng, chúng tôi đã tạo một cơ sở dữ liệu BioSQL mới và tải một số dữ liệu mẫu vào đó. Chúng ta sẽ thảo luận về các bảng quan trọng trong chương tiếp theo.
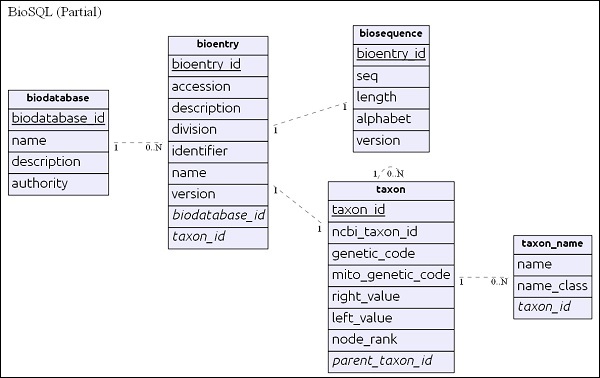
Sơ đồ ER đơn giản
biodatabase bảng nằm trên cùng của hệ thống phân cấp và mục đích chính của nó là tổ chức một tập hợp dữ liệu trình tự thành một nhóm / cơ sở dữ liệu ảo duy nhất. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. Tất cả các bảng liên quan trong cơ sở dữ liệu BioSQL đều có tham chiếu đến mục nhập cơ sở dữ liệu sinh học.
bioentrybảng chứa tất cả các chi tiết về một chuỗi ngoại trừ dữ liệu chuỗi. dữ liệu trình tự của một cụ thểbioentry sẽ được lưu trữ trong biosequence bàn.
taxon và taxon_name là các chi tiết phân loại và mọi mục nhập đều tham chiếu đến bảng này để chỉ định thông tin về đơn vị phân loại của nó.
Sau khi hiểu lược đồ, chúng ta hãy xem xét một số truy vấn trong phần tiếp theo.
Truy vấn BioSQL
Chúng ta hãy đi sâu vào một số truy vấn SQL để hiểu rõ hơn cách dữ liệu được tổ chức và các bảng có liên quan với nhau. Trước khi tiếp tục, chúng ta hãy mở cơ sở dữ liệu bằng lệnh dưới đây và đặt một số lệnh định dạng -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Bạn cũng có thể sử dụng bất kỳ trình soạn thảo SQLite nào để chạy truy vấn.
Liệt kê cơ sở dữ liệu chuỗi ảo có sẵn trong hệ thống như dưới đây:
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Ở đây, chúng tôi chỉ có một cơ sở dữ liệu, orchid.
Liệt kê các mục (đầu trang 3) có sẵn trong cơ sở dữ liệu orchid với mã cho sẵn dưới đây
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Liệt kê chi tiết trình tự được liên kết với mục nhập (gia nhập - Z78530, tên - gen C. fasciculatum 5.8S rRNA và DNA ITS1 và ITS2) với mã đã cho -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Nhận trình tự hoàn chỉnh được liên kết với mục nhập (gia nhập - Z78530, tên - gen C. fasciculatum 5.8S rRNA và DNA ITS1 và ITS2) bằng cách sử dụng mã dưới đây -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Liệt kê đơn vị phân loại liên kết với cơ sở dữ liệu sinh học, phong lan
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>Tải dữ liệu vào cơ sở dữ liệu BioSQL
Hãy để chúng tôi tìm hiểu cách tải dữ liệu tuần tự vào cơ sở dữ liệu BioSQL trong chương này. Chúng tôi đã có mã để tải dữ liệu vào cơ sở dữ liệu trong phần trước và mã như sau:
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Chúng ta sẽ có cái nhìn sâu hơn về từng dòng mã và mục đích của nó -
Line 1 - Tải mô-đun SeqIO.
Line 2- Tải mô-đun BioSeqDatabase. Mô-đun này cung cấp tất cả các chức năng để tương tác với cơ sở dữ liệu BioSQL.
Line 3 - Tải mô-đun os.
Line 5- open_database mở cơ sở dữ liệu được chỉ định (db) với trình điều khiển (trình điều khiển) được cấu hình và trả về một xử lý cho cơ sở dữ liệu BioSQL (máy chủ). Biopython hỗ trợ cơ sở dữ liệu sqlite, mysql, postgresql và oracle.
Line 6-10- Phương thức load_database_sql tải sql từ tệp bên ngoài và thực thi nó. phương thức cam kết thực hiện giao dịch. Chúng ta có thể bỏ qua bước này vì chúng ta đã tạo cơ sở dữ liệu bằng lược đồ.
Line 12 - Phương thức new_database tạo cơ sở dữ liệu ảo mới, lan và trả về một db xử lý để thực thi lệnh đối với cơ sở dữ liệu lan.
Line 13- load method tải các mục trình tự (SeqRecord có thể lặp lại) vào cơ sở dữ liệu lan. SqlIO.parse phân tích cú pháp cơ sở dữ liệu GenBank và trả về tất cả các chuỗi trong đó dưới dạng SeqRecord có thể lặp lại. Tham số thứ hai (True) của phương thức tải hướng dẫn nó tìm nạp các chi tiết phân loại của dữ liệu trình tự từ trang web nổ NCBI, nếu nó chưa có sẵn trong hệ thống.
Line 14 - cam kết cam kết giao dịch.
Line 15 - Close đóng kết nối cơ sở dữ liệu và hủy bỏ xử lý máy chủ.
Tìm nạp dữ liệu trình tự
Hãy để chúng tôi tìm nạp một chuỗi với số nhận dạng, 2765658 từ cơ sở dữ liệu phong lan như bên dưới -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Ở đây, máy chủ ["Orchid"] trả về xử lý để tìm nạp dữ liệu từ cơ sở dữ liệu ảo. lookup phương pháp cung cấp một tùy chọn để chọn trình tự dựa trên tiêu chí và chúng tôi đã chọn trình tự có số nhận dạng, 2765658. lookuptrả về thông tin trình tự dưới dạng SeqRecordobject. Vì chúng tôi đã biết cách làm việc với SeqRecord`, nên việc lấy dữ liệu từ nó rất dễ dàng.
Xóa cơ sở dữ liệu
Việc xóa cơ sở dữ liệu đơn giản như gọi phương thức remove_database với tên cơ sở dữ liệu thích hợp và sau đó cam kết nó như được chỉ định bên dưới:
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()