Biopython - Hướng dẫn nhanh
Biopython là gói tin sinh học lớn nhất và phổ biến nhất dành cho Python. Nó chứa một số mô-đun con khác nhau cho các nhiệm vụ tin sinh học thông thường. Nó được phát triển bởi Chapman và Chang, chủ yếu được viết bằng Python. Nó cũng chứa mã C để tối ưu hóa phần tính toán phức tạp của phần mềm. Nó chạy trên Windows, Linux, Mac OS X, v.v.
Về cơ bản, Biopython là một tập hợp các mô-đun python cung cấp các chức năng để xử lý các hoạt động của chuỗi DNA, RNA và protein như bổ sung ngược lại chuỗi DNA, tìm các mô típ trong chuỗi protein, v.v. Nó cung cấp rất nhiều trình phân tích cú pháp để đọc tất cả các cơ sở dữ liệu di truyền chính. như GenBank, SwissPort, FASTA, v.v., cũng như các trình bao bọc / giao diện để chạy phần mềm / công cụ tin sinh học phổ biến khác như NCBI BLASTN, Entrez, v.v., bên trong môi trường python. Nó có các dự án anh em như BioPerl, BioJava và BioRuby.
Đặc trưng
Biopython có tính di động, rõ ràng và có cú pháp dễ học. Một số tính năng nổi bật được liệt kê dưới đây:
Phiên dịch, tương tác và hướng đối tượng.
Hỗ trợ các định dạng liên quan đến FASTA, PDB, GenBank, Blast, SCOP, PubMed / Medline, ExPASy.
Tùy chọn để đối phó với các định dạng trình tự.
Các công cụ để quản lý cấu trúc protein.
BioSQL - Bộ bảng SQL tiêu chuẩn để lưu trữ chuỗi cộng với các tính năng và chú thích.
Truy cập vào các dịch vụ trực tuyến và cơ sở dữ liệu, bao gồm các dịch vụ NCBI (Blast, Entrez, PubMed) và các dịch vụ ExPASY (SwissProt, Prosite).
Truy cập vào các dịch vụ địa phương, bao gồm Blast, Clustalw, EMBOSS.
Bàn thắng
Mục tiêu của Biopython là cung cấp quyền truy cập đơn giản, tiêu chuẩn và rộng rãi vào tin sinh học thông qua ngôn ngữ python. Các mục tiêu cụ thể của Biopython được liệt kê dưới đây:
Cung cấp quyền truy cập tiêu chuẩn vào các nguồn tin sinh học.
Các mô-đun và tập lệnh chất lượng cao, có thể tái sử dụng.
Thao tác mảng nhanh có thể được sử dụng trong mã Cụm, PDB, NaiveBayes và Mô hình Markov.
Phân tích dữ liệu bộ gen.
Ưu điểm
Biopython yêu cầu rất ít mã và có những ưu điểm sau:
Cung cấp kiểu dữ liệu microarray được sử dụng trong phân cụm.
Đọc và ghi các tệp kiểu Tree-View.
Hỗ trợ dữ liệu cấu trúc được sử dụng để phân tích cú pháp, biểu diễn và phân tích PDB.
Hỗ trợ dữ liệu tạp chí được sử dụng trong các ứng dụng Medline.
Hỗ trợ cơ sở dữ liệu BioSQL, đây là cơ sở dữ liệu tiêu chuẩn được sử dụng rộng rãi trong số tất cả các dự án tin sinh học.
Hỗ trợ phát triển trình phân tích cú pháp bằng cách cung cấp các mô-đun để phân tích cú pháp tệp tin sinh học thành một đối tượng bản ghi có định dạng cụ thể hoặc một lớp trình tự cộng với các tính năng chung.
Tài liệu rõ ràng dựa trên kiểu sách nấu ăn.
Nghiên cứu điển hình mẫu
Hãy để chúng tôi kiểm tra một số trường hợp sử dụng (di truyền quần thể, cấu trúc RNA, v.v.) và cố gắng hiểu Biopython đóng vai trò quan trọng như thế nào trong lĩnh vực này -
Di truyền dân số
Di truyền quần thể là nghiên cứu về sự biến đổi di truyền trong một quần thể và liên quan đến việc kiểm tra và mô hình hóa những thay đổi về tần số của gen và alen trong quần thể theo không gian và thời gian.
Biopython cung cấp mô-đun Bio.PopGen cho di truyền quần thể. Mô-đun này chứa tất cả các chức năng cần thiết để thu thập thông tin về di truyền quần thể cổ điển.
Cấu trúc RNA
Ba đại phân tử sinh học chính cần thiết cho sự sống của chúng ta là DNA, RNA và Protein. Protein là con ngựa của tế bào và đóng một vai trò quan trọng như các enzym. DNA (axit deoxyribonucleic) được coi là “bản thiết kế” của tế bào. Nó mang tất cả thông tin di truyền cần thiết để tế bào phát triển, lấy chất dinh dưỡng và nhân giống. RNA (axit Ribonucleic) hoạt động như "bản sao DNA" trong tế bào.
Biopython cung cấp các đối tượng Bio.Sequence đại diện cho các nucleotide, các khối xây dựng DNA và RNA.
Phần này giải thích cách cài đặt Biopython trên máy tính của bạn. Nó rất dễ cài đặt và sẽ không mất quá năm phút.
Step 1 - Xác minh cài đặt Python
Biopython được thiết kế để hoạt động với Python 2.5 hoặc các phiên bản cao hơn. Vì vậy, bắt buộc phải cài đặt python trước. Chạy lệnh dưới đây trong dấu nhắc lệnh của bạn -
> python --versionNó được định nghĩa dưới đây -
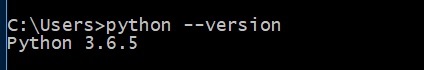
Nó hiển thị phiên bản của python, nếu được cài đặt đúng cách. Nếu không, hãy tải xuống phiên bản python mới nhất, cài đặt nó và sau đó chạy lại lệnh.
Step 2 - Cài đặt Biopython bằng pip
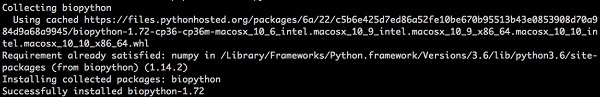
Dễ dàng cài đặt Biopython bằng cách sử dụng pip từ dòng lệnh trên tất cả các nền tảng. Nhập lệnh dưới đây -
> pip install biopythonPhản hồi sau sẽ được hiển thị trên màn hình của bạn -
Để cập nhật phiên bản cũ hơn của Biopython -
> pip install biopython –-upgradePhản hồi sau sẽ được hiển thị trên màn hình của bạn -
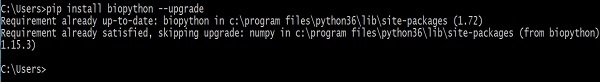
Sau khi thực hiện lệnh này, các phiên bản cũ hơn của Biopython và NumPy (tùy thuộc vào Biopython) sẽ bị xóa trước khi cài đặt các phiên bản gần đây.
Step 3 - Xác minh cài đặt Biopython
Bây giờ, bạn đã cài đặt thành công Biopython trên máy của mình. Để xác minh rằng Biopython được cài đặt đúng cách, hãy nhập lệnh dưới đây trên bảng điều khiển python của bạn -
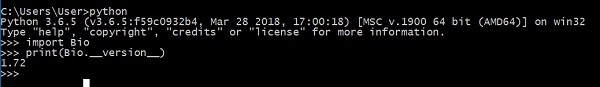
Nó hiển thị phiên bản của Biopython.
Alternate Way − Installing Biopython using Source
Để cài đặt Biopython bằng mã nguồn, hãy làm theo hướng dẫn sau:
Tải xuống bản phát hành gần đây của Biopython từ liên kết sau: https://biopython.org/wiki/Download
Hiện tại, phiên bản mới nhất là biopython-1.72.
Tải xuống tệp và giải nén tệp lưu trữ đã nén, chuyển vào thư mục mã nguồn và nhập lệnh dưới đây:
> python setup.py buildĐiều này sẽ xây dựng Biopython từ mã nguồn như dưới đây:
Bây giờ, hãy kiểm tra mã bằng lệnh dưới đây:
> python setup.py test
Cuối cùng, cài đặt bằng lệnh dưới đây -
> python setup.py install
Hãy để chúng tôi tạo một ứng dụng Biopython đơn giản để phân tích cú pháp tệp tin sinh học và in nội dung. Điều này sẽ giúp chúng ta hiểu được khái niệm chung về Biopython và nó giúp ích như thế nào trong lĩnh vực tin sinh học.
Step 1 - Đầu tiên, tạo một tệp trình tự mẫu, “example.fasta” và đưa nội dung bên dưới vào đó.
>sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAV
NNFEAHTINTVVHTNDSDKGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITID
SNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTAGQYQGLVSIILTKSTTTTTTTKGT
>sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVS
NTLVGVLTLSNTSIDTVSIASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDK
NAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGNYRANITITSTIKGGGTKKGTTDKKPhần mở rộng, fasta đề cập đến định dạng tệp của tệp trình tự. FASTA bắt nguồn từ phần mềm tin sinh học, FASTA và do đó nó có tên như vậy. Định dạng FASTA có nhiều chuỗi được sắp xếp từng cái một và mỗi chuỗi sẽ có id, tên, mô tả và dữ liệu chuỗi thực tế của riêng nó.
Step 2 - Tạo một tập lệnh python mới, * simple_example.py "và nhập mã bên dưới và lưu nó.
from Bio.SeqIO import parse
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
file = open("example.fasta")
records = parse(file, "fasta") for record in records:
print("Id: %s" % record.id)
print("Name: %s" % record.name)
print("Description: %s" % record.description)
print("Annotations: %s" % record.annotations)
print("Sequence Data: %s" % record.seq)
print("Sequence Alphabet: %s" % record.seq.alphabet)Hãy để chúng tôi xem xét sâu hơn một chút về mã -
Line 1nhập lớp phân tích cú pháp có sẵn trong mô-đun Bio.SeqIO. Mô-đun Bio.SeqIO được sử dụng để đọc và ghi tệp trình tự ở định dạng khác nhau và lớp phân tích cú pháp được sử dụng để phân tích nội dung của tệp trình tự.
Line 2nhập lớp SeqRecord có sẵn trong mô-đun Bio.SeqRecord. Mô-đun này được sử dụng để thao tác với các bản ghi trình tự và lớp SeqRecord được sử dụng để biểu diễn một trình tự cụ thể có sẵn trong tệp trình tự.
*Line 3"nhập lớp Seq có sẵn trong mô-đun Bio.Seq. Mô-đun này được sử dụng để thao tác dữ liệu trình tự và lớp Seq được sử dụng để biểu diễn dữ liệu trình tự của một bản ghi trình tự cụ thể có sẵn trong tệp trình tự.
Line 5 mở tệp “example.fasta” bằng cách sử dụng hàm python thông thường, mở.
Line 7 phân tích cú pháp nội dung của tệp trình tự và trả về nội dung dưới dạng danh sách đối tượng SeqRecord.
Line 9-15 lặp qua các bản ghi bằng cách sử dụng vòng lặp python for và in các thuộc tính của bản ghi trình tự (SqlRecord) như id, tên, mô tả, dữ liệu trình tự, v.v.
Line 15 in kiểu của dãy bằng cách sử dụng lớp Bảng chữ cái.
Step 3 - Mở dấu nhắc lệnh và đi đến thư mục chứa tệp trình tự, “example.fasta” và chạy lệnh dưới đây -
> python simple_example.pyStep 4- Python chạy tập lệnh và in tất cả dữ liệu trình tự có sẵn trong tệp mẫu, “example.fasta”. Kết quả đầu ra sẽ tương tự như nội dung sau.
Id: sp|P25730|FMS1_ECOLI
Name: sp|P25730|FMS1_ECOLI
Decription: sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
Annotations: {}
Sequence Data: MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAVNNFEAHTINTVVHTNDSD
KGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITIDSNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTA
GQYQGLVSIILTKSTTTTTTTKGT
Sequence Alphabet: SingleLetterAlphabet()
Id: sp|P15488|FMS3_ECOLI
Name: sp|P15488|FMS3_ECOLI
Decription: sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
Annotations: {}
Sequence Data: MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVSNTLVGVLTLSNTSIDTVS
IASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDKNAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGN
YRANITITSTIKGGGTKKGTTDKK
Sequence Alphabet: SingleLetterAlphabet()Chúng ta đã thấy ba lớp, phân tích cú pháp, SeqRecord và Seq trong ví dụ này. Ba lớp này cung cấp hầu hết các chức năng và chúng ta sẽ tìm hiểu các lớp đó trong phần tới.
Trình tự là một loạt các chữ cái được sử dụng để đại diện cho protein, DNA hoặc RNA của một sinh vật. Nó được đại diện bởi lớp Seq. Lớp Seq được định nghĩa trong mô-đun Bio.Seq.
Hãy tạo một chuỗi đơn giản trong Biopython như hình dưới đây -
>>> from Bio.Seq import Seq
>>> seq = Seq("AGCT")
>>> seq
Seq('AGCT')
>>> print(seq)
AGCTỞ đây, chúng tôi đã tạo ra một chuỗi protein đơn giản AGCT và mỗi chữ cái đại diện cho Alanine, Glycine, Cysteine và Threonine.
Mỗi đối tượng Seq có hai thuộc tính quan trọng:
dữ liệu - chuỗi trình tự thực tế (AGCT)
bảng chữ cái - được sử dụng để thể hiện loại trình tự. ví dụ: trình tự DNA, trình tự RNA, v.v ... Theo mặc định, nó không đại diện cho bất kỳ trình tự nào và có tính chất chung chung.
Mô-đun bảng chữ cái
Các đối tượng Seq chứa thuộc tính Bảng chữ cái để chỉ định kiểu trình tự, các chữ cái và các phép toán có thể. Nó được định nghĩa trong mô-đun Bio.Alphabet. Bảng chữ cái có thể được định nghĩa như sau:
>>> from Bio.Seq import Seq
>>> myseq = Seq("AGCT")
>>> myseq
Seq('AGCT')
>>> myseq.alphabet
Alphabet()Mô-đun bảng chữ cái cung cấp các lớp bên dưới để đại diện cho các loại trình tự khác nhau. Bảng chữ cái - lớp cơ sở cho tất cả các loại bảng chữ cái.
SingleLetterAlphabet - Bảng chữ cái chung với các chữ cái có kích thước là một. Nó bắt nguồn từ Bảng chữ cái và tất cả các loại bảng chữ cái khác bắt nguồn từ nó.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import single_letter_alphabet
>>> test_seq = Seq('AGTACACTGGT', single_letter_alphabet)
>>> test_seq
Seq('AGTACACTGGT', SingleLetterAlphabet())ProteinAlphabet - Bảng chữ cái protein đơn chữ cái chung.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_protein
>>> test_seq = Seq('AGTACACTGGT', generic_protein)
>>> test_seq
Seq('AGTACACTGGT', ProteinAlphabet())NucleotideAlphabet - Bảng chữ cái nucleotide chung một chữ cái.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_nucleotide
>>> test_seq = Seq('AGTACACTGGT', generic_nucleotide) >>> test_seq
Seq('AGTACACTGGT', NucleotideAlphabet())DNAAlphabet - Bảng chữ cái DNA chữ cái chung.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_dna
>>> test_seq = Seq('AGTACACTGGT', generic_dna)
>>> test_seq
Seq('AGTACACTGGT', DNAAlphabet())RNAAlphabet - Bảng chữ cái RNA đơn chung chung.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_rna
>>> test_seq = Seq('AGTACACTGGT', generic_rna)
>>> test_seq
Seq('AGTACACTGGT', RNAAlphabet())Mô-đun Biopython, Bio.Alphabet.IUPAC cung cấp các loại trình tự cơ bản theo định nghĩa của cộng đồng IUPAC. Nó chứa các lớp sau:
IUPACProtein (protein) - Bảng chữ cái IUPAC protein gồm 20 loại axit amin tiêu chuẩn.
ExtendedIUPACProtein (extended_protein) - Bảng chữ cái đơn chữ cái viết hoa IUPAC viết hoa mở rộng bao gồm X.
IUPACAmbiguousDNA (ambiguous_dna) - IUPAC viết hoa không rõ ràng DNA.
IUPACUnambiguousDNA (unambiguous_dna) - IUPAC viết hoa rõ ràng DNA (GATC).
ExtendedIUPACDNA (extended_dna) - Bảng chữ cái IUPAC DNA mở rộng.
IUPACAmbiguousRNA (ambiguous_rna) - RNA mơ hồ IUPAC viết hoa.
IUPACUnambiguousRNA (unambiguous_rna) - Chữ hoa IUPAC RNA rõ ràng (GAUC).
Hãy xem xét một ví dụ đơn giản cho lớp IUPACProtein như hình dưới đây:
>>> from Bio.Alphabet import IUPAC
>>> protein_seq = Seq("AGCT", IUPAC.protein)
>>> protein_seq
Seq('AGCT', IUPACProtein())
>>> protein_seq.alphabetNgoài ra, Biopython hiển thị tất cả dữ liệu cấu hình liên quan đến tin sinh học thông qua mô-đun Bio.Data. Ví dụ: IUPACData.protein_letters có các chữ cái có thể có của bảng chữ cái IUPACProtein.
>>> from Bio.Data import IUPACData
>>> IUPACData.protein_letters
'ACDEFGHIKLMNPQRSTVWY'Hoạt động cơ bản
Phần này giải thích ngắn gọn về tất cả các thao tác cơ bản có sẵn trong lớp Seq. Các chuỗi tương tự như chuỗi python. Chúng tôi có thể thực hiện các hoạt động chuỗi python như cắt, đếm, nối, tìm, tách và dải theo chuỗi.
Sử dụng các mã dưới đây để nhận các đầu ra khác nhau.
To get the first value in sequence.
>>> seq_string = Seq("AGCTAGCT")
>>> seq_string[0]
'A'To print the first two values.
>>> seq_string[0:2]
Seq('AG')To print all the values.
>>> seq_string[ : ]
Seq('AGCTAGCT')To perform length and count operations.
>>> len(seq_string)
8
>>> seq_string.count('A')
2To add two sequences.
>>> from Bio.Alphabet import generic_dna, generic_protein
>>> seq1 = Seq("AGCT", generic_dna)
>>> seq2 = Seq("TCGA", generic_dna)
>>> seq1+seq2
Seq('AGCTTCGA', DNAAlphabet())Ở đây, hai đối tượng trình tự ở trên, seq1, seq2 là trình tự DNA chung và do đó bạn có thể thêm chúng và tạo ra trình tự mới. Bạn không thể thêm các trình tự có bảng chữ cái không tương thích, chẳng hạn như trình tự protein và trình tự DNA như được chỉ định bên dưới -
>>> dna_seq = Seq('AGTACACTGGT', generic_dna)
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> dna_seq + protein_seq
.....
.....
TypeError: Incompatible alphabets DNAAlphabet() and ProteinAlphabet()
>>>Để thêm hai hoặc nhiều chuỗi, trước tiên hãy lưu trữ nó trong một danh sách python, sau đó truy xuất nó bằng 'vòng lặp for' và cuối cùng thêm nó lại với nhau như được hiển thị bên dưới:
>>> from Bio.Alphabet import generic_dna
>>> list = [Seq("AGCT",generic_dna),Seq("TCGA",generic_dna),Seq("AAA",generic_dna)]
>>> for s in list:
... print(s)
...
AGCT
TCGA
AAA
>>> final_seq = Seq(" ",generic_dna)
>>> for s in list:
... final_seq = final_seq + s
...
>>> final_seq
Seq('AGCTTCGAAAA', DNAAlphabet())Trong phần dưới đây, các mã khác nhau được đưa ra để nhận kết quả đầu ra dựa trên yêu cầu.
To change the case of sequence.
>>> from Bio.Alphabet import generic_rna
>>> rna = Seq("agct", generic_rna)
>>> rna.upper()
Seq('AGCT', RNAAlphabet())To check python membership and identity operator.
>>> rna = Seq("agct", generic_rna)
>>> 'a' in rna
True
>>> 'A' in rna
False
>>> rna1 = Seq("AGCT", generic_dna)
>>> rna is rna1
FalseTo find single letter or sequence of letter inside the given sequence.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.find('G')
1
>>> protein_seq.find('GG')
8To perform splitting operation.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.split('A')
[Seq('', ProteinAlphabet()), Seq('GU', ProteinAlphabet()),
Seq('C', ProteinAlphabet()), Seq('CUGGU', ProteinAlphabet())]To perform strip operations in the sequence.
>>> strip_seq = Seq(" AGCT ")
>>> strip_seq
Seq(' AGCT ')
>>> strip_seq.strip()
Seq('AGCT')Trong chương này, chúng ta sẽ thảo luận về một số tính năng trình tự nâng cao do Biopython cung cấp.
Bổ sung và Bổ sung ngược
Trình tự nucleotide có thể được bổ sung ngược lại để có được trình tự mới. Ngoài ra, trình tự bổ sung có thể được bổ sung ngược lại để có được trình tự ban đầu. Biopython cung cấp hai phương pháp để thực hiện chức năng này -complement và reverse_complement. Mã cho điều này được cung cấp dưới đây:
>>> from Bio.Alphabet import IUPAC
>>> nucleotide = Seq('TCGAAGTCAGTC', IUPAC.ambiguous_dna)
>>> nucleotide.complement()
Seq('AGCTTCAGTCAG', IUPACAmbiguousDNA())
>>>Ở đây, phương thức bổ sung () cho phép bổ sung một chuỗi DNA hoặc RNA. Phương thức reverse_complement () bổ sung và đảo ngược chuỗi kết quả từ trái sang phải. Nó được hiển thị bên dưới -
>>> nucleotide.reverse_complement()
Seq('GACTGACTTCGA', IUPACAmbiguousDNA())Biopython sử dụng biến mập mờ_dna_complement do Bio.Data.IUPACData cung cấp để thực hiện hoạt động bổ sung.
>>> from Bio.Data import IUPACData
>>> import pprint
>>> pprint.pprint(IUPACData.ambiguous_dna_complement) {
'A': 'T',
'B': 'V',
'C': 'G',
'D': 'H',
'G': 'C',
'H': 'D',
'K': 'M',
'M': 'K',
'N': 'N',
'R': 'Y',
'S': 'S',
'T': 'A',
'V': 'B',
'W': 'W',
'X': 'X',
'Y': 'R'}
>>>Nội dung GC
Thành phần cơ sở DNA bộ gen (hàm lượng GC) được dự đoán sẽ ảnh hưởng đáng kể đến hoạt động của bộ gen và sinh thái loài. Hàm lượng GC là số lượng nucleotide GC chia cho tổng số nucleotide.
Để nhận nội dung nucleotide GC, hãy nhập mô-đun sau và thực hiện các bước sau:
>>> from Bio.SeqUtils import GC
>>> nucleotide = Seq("GACTGACTTCGA",IUPAC.unambiguous_dna)
>>> GC(nucleotide)
50.0Phiên mã
Phiên mã là quá trình thay đổi chuỗi DNA thành chuỗi RNA. Quá trình phiên mã sinh học thực tế đang thực hiện bổ sung ngược (TCAG → CUGA) để lấy mRNA coi DNA là sợi khuôn. Tuy nhiên, trong tin sinh học và trong Biopython cũng vậy, chúng tôi thường làm việc trực tiếp với chuỗi mã hóa và chúng tôi có thể nhận được trình tự mRNA bằng cách thay đổi chữ T thành U.
Ví dụ đơn giản cho phần trên như sau:
>>> from Bio.Seq import Seq
>>> from Bio.Seq import transcribe
>>> from Bio.Alphabet import IUPAC
>>> dna_seq = Seq("ATGCCGATCGTAT",IUPAC.unambiguous_dna) >>> transcribe(dna_seq)
Seq('AUGCCGAUCGUAU', IUPACUnambiguousRNA())
>>>Để đảo ngược phiên mã, T được đổi thành U như trong đoạn mã dưới đây -
>>> rna_seq = transcribe(dna_seq)
>>> rna_seq.back_transcribe()
Seq('ATGCCGATCGTAT', IUPACUnambiguousDNA())Để lấy chuỗi khuôn mẫu DNA, hãy đảo ngược_complement RNA được phiên mã ngược như được đưa ra bên dưới:
>>> rna_seq.back_transcribe().reverse_complement()
Seq('ATACGATCGGCAT', IUPACUnambiguousDNA())Dịch
Dịch mã là một quá trình phiên mã chuỗi RNA sang chuỗi protein. Hãy xem xét một chuỗi RNA như hình dưới đây:
>>> rna_seq = Seq("AUGGCCAUUGUAAU",IUPAC.unambiguous_rna)
>>> rna_seq
Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())Bây giờ, hãy áp dụng hàm translate () cho đoạn mã trên -
>>> rna_seq.translate()
Seq('MAIV', IUPACProtein())Trình tự RNA trên là đơn giản. Xem xét trình tự RNA, AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA và áp dụng dịch () -
>>> rna = Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA', IUPAC.unambiguous_rna)
>>> rna.translate()
Seq('MAIVMGR*KGAR', HasStopCodon(IUPACProtein(), '*'))Tại đây, các mã dừng được biểu thị bằng dấu sao '*'.
Trong phương thức translate () có thể dừng ở codon dừng đầu tiên. Để thực hiện việc này, bạn có thể gán to_stop = True trong translate () như sau:
>>> rna.translate(to_stop = True)
Seq('MAIVMGR', IUPACProtein())Ở đây, codon dừng không được bao gồm trong trình tự kết quả vì nó không chứa mã này.
Bảng dịch
Trang Mã di truyền của NCBI cung cấp danh sách đầy đủ các bảng dịch được Biopython sử dụng. Hãy để chúng tôi xem một ví dụ cho bảng tiêu chuẩn để hình dung mã -
>>> from Bio.Data import CodonTable
>>> table = CodonTable.unambiguous_dna_by_name["Standard"]
>>> print(table)
Table 1 Standard, SGC0
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--
>>>Biopython sử dụng bảng này để dịch DNA thành protein cũng như tìm mã số Stop.
Biopython cung cấp một mô-đun, Bio.SeqIO để đọc và ghi các trình tự từ và vào một tệp (bất kỳ luồng nào) tương ứng. Nó hỗ trợ gần như tất cả các định dạng tệp có sẵn trong tin sinh học. Hầu hết các phần mềm cung cấp cách tiếp cận khác nhau cho các định dạng tệp khác nhau. Tuy nhiên, Biopython tuân theo một cách có ý thức một cách tiếp cận duy nhất để trình bày dữ liệu trình tự đã được phân tích cú pháp cho người dùng thông qua đối tượng SeqRecord của nó.
Hãy cùng chúng tôi tìm hiểu thêm về SeqRecord trong phần sau.
SeqRecord
Mô-đun Bio.SeqRecord cung cấp SeqRecord để giữ thông tin meta của chuỗi cũng như dữ liệu chuỗi như được cung cấp bên dưới:
seq - Đó là một chuỗi thực tế.
id - Nó là định danh chính của dãy đã cho. Loại mặc định là chuỗi.
tên - Nó là Tên của dãy. Loại mặc định là chuỗi.
mô tả - Nó hiển thị thông tin con người có thể đọc được về trình tự.
chú thích - Nó là một từ điển thông tin bổ sung về trình tự.
SeqRecord có thể được nhập như được chỉ định bên dưới
from Bio.SeqRecord import SeqRecordHãy để chúng tôi hiểu các sắc thái của việc phân tích cú pháp tệp trình tự bằng tệp trình tự thực trong các phần tiếp theo.
Phân tích cú pháp các định dạng tệp trình tự
Phần này giải thích về cách phân tích cú pháp hai trong số các định dạng tệp trình tự phổ biến nhất, FASTA và GenBank.
FASTA
FASTAlà định dạng tệp cơ bản nhất để lưu trữ dữ liệu trình tự. Ban đầu, FASTA là một gói phần mềm để sắp xếp trình tự của DNA và protein được phát triển trong quá trình phát triển ban đầu của Tin sinh học và được sử dụng chủ yếu để tìm kiếm sự tương đồng về trình tự.
Biopython cung cấp một tệp FASTA mẫu và nó có thể được truy cập tại https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Tải xuống và lưu tệp này vào thư mục mẫu Biopython của bạn dưới dạng ‘orchid.fasta’.
Mô-đun Bio.SeqIO cung cấp phương thức parse () để xử lý các tệp trình tự và có thể được nhập như sau:
from Bio.SeqIO import parsePhương thức parse () chứa hai đối số, đối số đầu tiên là tệp xử lý và đối số thứ hai là định dạng tệp.
>>> file = open('path/to/biopython/sample/orchid.fasta')
>>> for record in parse(file, "fasta"):
... print(record.id)
...
gi|2765658|emb|Z78533.1|CIZ78533
gi|2765657|emb|Z78532.1|CCZ78532
..........
..........
gi|2765565|emb|Z78440.1|PPZ78440
gi|2765564|emb|Z78439.1|PBZ78439
>>>Ở đây, phương thức parse () trả về một đối tượng có thể lặp lại, nó trả về SeqRecord trên mỗi lần lặp. Có thể lặp lại, nó cung cấp nhiều phương pháp phức tạp và dễ dàng và cho chúng ta xem một số tính năng.
kế tiếp()
Phương thức next () trả về mục tiếp theo có sẵn trong đối tượng có thể lặp lại, chúng ta có thể sử dụng mục này để lấy chuỗi đầu tiên như được cho bên dưới:
>>> first_seq_record = next(SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta'))
>>> first_seq_record.id 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.name 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', SingleLetterAlphabet())
>>> first_seq_record.description 'gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> first_seq_record.annotations
{}
>>>Ở đây, seq_record.annotations trống vì định dạng FASTA không hỗ trợ chú thích trình tự.
danh sách hiểu
Chúng tôi có thể chuyển đổi đối tượng có thể lặp lại thành danh sách bằng cách sử dụng tính năng hiểu danh sách như được cung cấp bên dưới
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> all_seq = [seq_record for seq_record in seq_iter] >>> len(all_seq)
94
>>>Ở đây, chúng tôi đã sử dụng phương thức len để lấy tổng số. Chúng ta có thể nhận được chuỗi có độ dài tối đa như sau:
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> max_seq = max(len(seq_record.seq) for seq_record in seq_iter)
>>> max_seq
789
>>>Chúng tôi cũng có thể lọc trình tự bằng cách sử dụng mã dưới đây:
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> seq_under_600 = [seq_record for seq_record in seq_iter if len(seq_record.seq) < 600]
>>> for seq in seq_under_600:
... print(seq.id)
...
gi|2765606|emb|Z78481.1|PIZ78481
gi|2765605|emb|Z78480.1|PGZ78480
gi|2765601|emb|Z78476.1|PGZ78476
gi|2765595|emb|Z78470.1|PPZ78470
gi|2765594|emb|Z78469.1|PHZ78469
gi|2765564|emb|Z78439.1|PBZ78439
>>>Việc ghi tập hợp các đối tượng SqlRecord (dữ liệu được phân tích cú pháp) vào tệp đơn giản như cách gọi phương thức SeqIO.write như bên dưới:
file = open("converted.fasta", "w)
SeqIO.write(seq_record, file, "fasta")Phương pháp này có thể được sử dụng hiệu quả để chuyển đổi định dạng như được chỉ định bên dưới:
file = open("converted.gbk", "w)
SeqIO.write(seq_record, file, "genbank")GenBank
Nó là một định dạng trình tự phong phú hơn cho các gen và bao gồm các trường cho nhiều loại chú thích khác nhau. Biopython cung cấp một tệp GenBank mẫu và nó có thể được truy cập tạihttps://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Tải xuống và lưu tệp vào thư mục mẫu Biopython của bạn dưới dạng ‘orchid.gbk’
Kể từ đó, Biopython cung cấp một chức năng duy nhất, phân tích cú pháp để phân tích cú pháp tất cả các định dạng tin sinh học. Phân tích cú pháp định dạng GenBank đơn giản như thay đổi tùy chọn định dạng trong phương pháp phân tích cú pháp.
Mã cho điều tương tự đã được đưa ra bên dưới -
>>> from Bio import SeqIO
>>> from Bio.SeqIO import parse
>>> seq_record = next(parse(open('path/to/biopython/sample/orchid.gbk'),'genbank'))
>>> seq_record.id
'Z78533.1'
>>> seq_record.name
'Z78533'
>>> seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', IUPACAmbiguousDNA())
>>> seq_record.description
'C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> seq_record.annotations {
'molecule_type': 'DNA',
'topology': 'linear',
'data_file_division': 'PLN',
'date': '30-NOV-2006',
'accessions': ['Z78533'],
'sequence_version': 1,
'gi': '2765658',
'keywords': ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'],
'source': 'Cypripedium irapeanum',
'organism': 'Cypripedium irapeanum',
'taxonomy': [
'Eukaryota',
'Viridiplantae',
'Streptophyta',
'Embryophyta',
'Tracheophyta',
'Spermatophyta',
'Magnoliophyta',
'Liliopsida',
'Asparagales',
'Orchidaceae',
'Cypripedioideae',
'Cypripedium'],
'references': [
Reference(title = 'Phylogenetics of the slipper orchids (Cypripedioideae:
Orchidaceae): nuclear rDNA ITS sequences', ...),
Reference(title = 'Direct Submission', ...)
]
}Sequence alignment là quá trình sắp xếp hai hay nhiều trình tự (của trình tự DNA, RNA hoặc protein) theo một trật tự cụ thể để xác định vùng tương đồng giữa chúng.
Việc xác định vùng tương tự cho phép chúng ta suy ra nhiều thông tin như những đặc điểm nào được bảo tồn giữa các loài, các loài khác nhau gần gũi về mặt di truyền như thế nào, các loài tiến hóa như thế nào, v.v. Biopython cung cấp hỗ trợ rộng rãi cho việc sắp xếp trình tự.
Hãy để chúng tôi tìm hiểu một số tính năng quan trọng được cung cấp bởi Biopython trong chương này -
Phân tích cú pháp sắp xếp trình tự
Biopython cung cấp một mô-đun, Bio.AlignIO để đọc và ghi các liên kết trình tự. Trong tin sinh học, có rất nhiều định dạng có sẵn để chỉ định dữ liệu sắp xếp trình tự tương tự như dữ liệu trình tự đã học trước đó. Bio.AlignIO cung cấp API tương tự như Bio.SeqIO ngoại trừ việc Bio.SeqIO hoạt động trên dữ liệu trình tự và Bio.AlignIO hoạt động trên dữ liệu căn chỉnh trình tự.
Trước khi bắt đầu tìm hiểu, chúng ta hãy tải xuống tệp căn chỉnh trình tự mẫu từ Internet.
Để tải xuống tệp mẫu, hãy làm theo các bước sau:
Step 1 - Mở trình duyệt yêu thích của bạn và truy cập http://pfam.xfam.org/family/browsetrang mạng. Nó sẽ hiển thị tất cả các họ Pfam theo thứ tự bảng chữ cái.
Step 2- Chọn một họ bất kỳ có số lượng hạt giống ít hơn. Nó chứa dữ liệu tối thiểu và cho phép chúng tôi làm việc dễ dàng với sự liên kết. Ở đây, chúng tôi đã chọn / nhấp vào PF18225 và nó sẽ mở rahttp://pfam.xfam.org/family/PF18225 và hiển thị chi tiết đầy đủ về nó, bao gồm cả căn chỉnh trình tự.
Step 3 - Vào phần căn chỉnh và tải xuống tệp căn chỉnh trình tự ở định dạng Stockholm (PF18225_seed.txt).
Chúng ta hãy thử đọc tệp căn chỉnh trình tự đã tải xuống bằng Bio.AlignIO như bên dưới -
Nhập mô-đun Bio.AlignIO
>>> from Bio import AlignIOĐọc căn chỉnh bằng phương pháp đọc. phương thức đọc được sử dụng để đọc dữ liệu căn chỉnh đơn có sẵn trong tệp đã cho. Nếu tệp đã cho chứa nhiều căn chỉnh, chúng ta có thể sử dụng phương pháp phân tích cú pháp. phương thức phân tích cú pháp trả về đối tượng căn chỉnh có thể lặp lại tương tự như phương thức phân tích cú pháp trong mô-đun Bio.SeqIO.
>>> alignment = AlignIO.read(open("PF18225_seed.txt"), "stockholm")In đối tượng căn chỉnh.
>>> print(alignment)
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Chúng tôi cũng có thể kiểm tra các chuỗi (SeqRecord) có sẵn trong căn chỉnh cũng như bên dưới -
>>> for align in alignment:
... print(align.seq)
...
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVATVANQLRGRKRRAFARHREGP
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADITA---RLDRRREHGEHGVRKKP
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMAPMLIALNYRNRESHAQVDKKP
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMAPLFKVLSFRNREDQGLVNNKP
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIMVLAPRLTAKHPYDKVQDRNRK
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVADLMRKLDLDRPFKKLERKNRT
>>>Nhiều căn chỉnh
Nói chung, hầu hết các tệp căn chỉnh trình tự đều chứa dữ liệu căn chỉnh duy nhất và nó đủ để sử dụng readđể phân tích cú pháp nó. Trong khái niệm căn chỉnh nhiều trình tự, hai hoặc nhiều trình tự được so sánh để có các kết quả phù hợp thứ tự tốt nhất giữa chúng và dẫn đến nhiều trình tự căn chỉnh trong một tệp duy nhất.
Nếu định dạng căn chỉnh trình tự đầu vào chứa nhiều hơn một căn chỉnh trình tự, thì chúng ta cần sử dụng parse phương pháp thay vì read như được chỉ định bên dưới -
>>> from Bio import AlignIO
>>> alignments = AlignIO.parse(open("PF18225_seed.txt"), "stockholm")
>>> print(alignments)
<generator object parse at 0x000001CD1C7E0360>
>>> for alignment in alignments:
... print(alignment)
...
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Ở đây, phương thức phân tích cú pháp trả về đối tượng căn chỉnh có thể lặp lại và nó có thể được lặp lại để có được căn chỉnh thực tế.
Căn chỉnh trình tự theo cặp
Pairwise sequence alignment chỉ so sánh hai trình tự cùng một lúc và cung cấp sự liên kết trình tự tốt nhất có thể. Pairwise dễ hiểu và đặc biệt để suy ra từ sự liên kết trình tự kết quả.
Biopython cung cấp một mô-đun đặc biệt, Bio.pairwise2để xác định trình tự căn chỉnh bằng phương pháp theo cặp. Biopython áp dụng thuật toán tốt nhất để tìm chuỗi căn chỉnh và nó ngang bằng với các phần mềm khác.
Hãy để chúng tôi viết một ví dụ để tìm sự liên kết trình tự của hai chuỗi đơn giản và giả thuyết bằng cách sử dụng mô-đun ghép đôi. Điều này sẽ giúp chúng ta hiểu khái niệm về căn chỉnh trình tự và cách lập trình nó bằng Biopython.
Bước 1
Nhập mô-đun pairwise2 với lệnh dưới đây -
>>> from Bio import pairwise2Bước 2
Tạo hai chuỗi, seq1 và seq2 -
>>> from Bio.Seq import Seq
>>> seq1 = Seq("ACCGGT")
>>> seq2 = Seq("ACGT")Bước 3
Gọi phương thức pairwise2.align.globalxx cùng với seq1 và seq2 để tìm các căn chỉnh bằng cách sử dụng dòng mã bên dưới:
>>> alignments = pairwise2.align.globalxx(seq1, seq2)Đây, globalxxthực hiện công việc thực tế và tìm tất cả các liên kết tốt nhất có thể trong các trình tự nhất định. Trên thực tế, Bio.pairwise2 cung cấp khá nhiều phương pháp tuân theo quy ước dưới đây để tìm các căn chỉnh trong các tình huống khác nhau.
<sequence alignment type>XYỞ đây, kiểu căn chỉnh trình tự đề cập đến kiểu căn chỉnh có thể là toàn cục hoặc cục bộ. loại toàn cầu là tìm sự liên kết trình tự bằng cách xem xét toàn bộ trình tự. kiểu cục bộ là tìm sự liên kết trình tự bằng cách xem xét tập hợp con của các trình tự đã cho. Điều này sẽ tẻ nhạt nhưng cung cấp ý tưởng tốt hơn về sự giống nhau giữa các trình tự đã cho.
X là điểm phù hợp. Các giá trị có thể có là x (khớp chính xác), m (điểm dựa trên các ký tự giống hệt nhau), d (từ điển do người dùng cung cấp với ký tự và điểm khớp) và cuối cùng là c (hàm do người dùng xác định để cung cấp thuật toán chấm điểm tùy chỉnh).
Y đề cập đến hình phạt khoảng cách. Các giá trị có thể có là x (không có hình phạt khoảng cách), s (hình phạt giống nhau cho cả hai chuỗi), d (hình phạt khác nhau cho mỗi chuỗi) và cuối cùng là c (chức năng do người dùng xác định để cung cấp hình phạt khoảng cách tùy chỉnh)
Vì vậy, localds cũng là một phương pháp hợp lệ, tìm kiếm sự liên kết trình tự bằng cách sử dụng kỹ thuật căn chỉnh cục bộ, từ điển do người dùng cung cấp cho các trận đấu và hình phạt khoảng cách do người dùng cung cấp cho cả hai chuỗi.
>>> test_alignments = pairwise2.align.localds(seq1, seq2, blosum62, -10, -1)Ở đây, blosum62 đề cập đến một từ điển có sẵn trong mô-đun pairwise2 để cung cấp điểm đối sánh. -10 đề cập đến hình phạt mở khoảng cách và -1 là hình phạt mở rộng khoảng cách.
Bước 4
Lặp lại đối tượng căn chỉnh có thể lặp lại và lấy từng đối tượng căn chỉnh riêng lẻ và in nó.
>>> for alignment in alignments:
... print(alignment)
...
('ACCGGT', 'A-C-GT', 4.0, 0, 6)
('ACCGGT', 'AC--GT', 4.0, 0, 6)
('ACCGGT', 'A-CG-T', 4.0, 0, 6)
('ACCGGT', 'AC-G-T', 4.0, 0, 6)Bước 5
Mô-đun Bio.pairwise2 cung cấp một phương pháp định dạng, format_alignment để hình dung rõ hơn kết quả -
>>> from Bio.pairwise2 import format_alignment
>>> alignments = pairwise2.align.globalxx(seq1, seq2)
>>> for alignment in alignments:
... print(format_alignment(*alignment))
...
ACCGGT
| | ||
A-C-GT
Score=4
ACCGGT
|| ||
AC--GT
Score=4
ACCGGT
| || |
A-CG-T
Score=4
ACCGGT
|| | |
AC-G-T
Score=4
>>>Biopython cũng cung cấp một mô-đun khác để sắp xếp trình tự, Align. Mô-đun này cung cấp một bộ API khác để đơn giản là cài đặt tham số như thuật toán, chế độ, điểm trận đấu, hình phạt cách biệt, v.v., Cái nhìn đơn giản về đối tượng Align như sau:
>>> from Bio import Align
>>> aligner = Align.PairwiseAligner()
>>> print(aligner)
Pairwise sequence aligner with parameters
match score: 1.000000
mismatch score: 0.000000
target open gap score: 0.000000
target extend gap score: 0.000000
target left open gap score: 0.000000
target left extend gap score: 0.000000
target right open gap score: 0.000000
target right extend gap score: 0.000000
query open gap score: 0.000000
query extend gap score: 0.000000
query left open gap score: 0.000000
query left extend gap score: 0.000000
query right open gap score: 0.000000
query right extend gap score: 0.000000
mode: global
>>>Hỗ trợ cho các công cụ căn chỉnh trình tự
Biopython cung cấp giao diện cho rất nhiều công cụ căn chỉnh trình tự thông qua mô-đun Bio.Align.Application. Một số công cụ được liệt kê dưới đây:
- ClustalW
- MUSCLE
- Kim và nước EMBOSS
Hãy để chúng tôi viết một ví dụ đơn giản trong Biopython để tạo căn chỉnh trình tự thông qua công cụ căn chỉnh phổ biến nhất, ClustalW.
Step 1 - Tải xuống chương trình Clustalw từ http://www.clustal.org/download/current/và cài đặt nó. Ngoài ra, hãy cập nhật PATH hệ thống bằng đường dẫn cài đặt “clustal”.
Step 2 - nhập ClustalwCommanLine từ mô-đun Bio.Align.Application.
>>> from Bio.Align.Applications import ClustalwCommandlineStep 3 - Đặt cmd bằng cách gọi ClustalwCommanLine với tệp đầu vào, opuntia.fasta có sẵn trong gói Biopython. https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/opuntia.fasta
>>> cmd = ClustalwCommandline("clustalw2",
infile="/path/to/biopython/sample/opuntia.fasta")
>>> print(cmd)
clustalw2 -infile=fasta/opuntia.fastaStep 4 - Gọi cmd () sẽ chạy lệnh clustalw và đưa ra kết quả là tệp căn chỉnh kết quả, opuntia.aln.
>>> stdout, stderr = cmd()Step 5 - Đọc và in tệp căn chỉnh như bên dưới -
>>> from Bio import AlignIO
>>> align = AlignIO.read("/path/to/biopython/sample/opuntia.aln", "clustal")
>>> print(align)
SingleLetterAlphabet() alignment with 7 rows and 906 columns
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273285|gb|AF191659.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273284|gb|AF191658.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273287|gb|AF191661.1|AF191
TATACATAAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273286|gb|AF191660.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273290|gb|AF191664.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273289|gb|AF191663.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273291|gb|AF191665.1|AF191
>>>BLAST là viết tắt của Basic Local Alignment Search Tool. Nó tìm thấy các vùng tương đồng giữa các trình tự sinh học. Biopython cung cấp mô-đun Bio.Blast để đối phó với hoạt động NCBI BLAST. Bạn có thể chạy BLAST trong kết nối cục bộ hoặc qua kết nối Internet.
Hãy để chúng tôi hiểu ngắn gọn về hai kết nối này trong phần sau:
Chạy qua Internet
Biopython cung cấp mô-đun Bio.Blast.NCBIWWW để gọi phiên bản trực tuyến của BLAST. Để thực hiện việc này, chúng ta cần nhập mô-đun sau:
>>> from Bio.Blast import NCBIWWWMô-đun NCBIWW cung cấp chức năng qblast để truy vấn phiên bản trực tuyến BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi. qblast hỗ trợ tất cả các tham số được hỗ trợ bởi phiên bản trực tuyến.
Để nhận bất kỳ trợ giúp nào về mô-đun này, hãy sử dụng lệnh dưới đây và hiểu các tính năng -
>>> help(NCBIWWW.qblast)
Help on function qblast in module Bio.Blast.NCBIWWW:
qblast(
program, database, sequence,
url_base = 'https://blast.ncbi.nlm.nih.gov/Blast.cgi',
auto_format = None,
composition_based_statistics = None,
db_genetic_code = None,
endpoints = None,
entrez_query = '(none)',
expect = 10.0,
filter = None,
gapcosts = None,
genetic_code = None,
hitlist_size = 50,
i_thresh = None,
layout = None,
lcase_mask = None,
matrix_name = None,
nucl_penalty = None,
nucl_reward = None,
other_advanced = None,
perc_ident = None,
phi_pattern = None,
query_file = None,
query_believe_defline = None,
query_from = None,
query_to = None,
searchsp_eff = None,
service = None,
threshold = None,
ungapped_alignment = None,
word_size = None,
alignments = 500,
alignment_view = None,
descriptions = 500,
entrez_links_new_window = None,
expect_low = None,
expect_high = None,
format_entrez_query = None,
format_object = None,
format_type = 'XML',
ncbi_gi = None,
results_file = None,
show_overview = None,
megablast = None,
template_type = None,
template_length = None
)
BLAST search using NCBI's QBLAST server or a cloud service provider.
Supports all parameters of the qblast API for Put and Get.
Please note that BLAST on the cloud supports the NCBI-BLAST Common
URL API (http://ncbi.github.io/blast-cloud/dev/api.html).
To use this feature, please set url_base to 'http://host.my.cloud.service.provider.com/cgi-bin/blast.cgi' and
format_object = 'Alignment'. For more details, please see 8. Biopython – Overview of BLAST
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE = BlastDocs&DOC_TYPE = CloudBlast
Some useful parameters:
- program blastn, blastp, blastx, tblastn, or tblastx (lower case)
- database Which database to search against (e.g. "nr").
- sequence The sequence to search.
- ncbi_gi TRUE/FALSE whether to give 'gi' identifier.
- descriptions Number of descriptions to show. Def 500.
- alignments Number of alignments to show. Def 500.
- expect An expect value cutoff. Def 10.0.
- matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45).
- filter "none" turns off filtering. Default no filtering
- format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML".
- entrez_query Entrez query to limit Blast search
- hitlist_size Number of hits to return. Default 50
- megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only)
- service plain, psi, phi, rpsblast, megablast (lower case)
This function does no checking of the validity of the parameters
and passes the values to the server as is. More help is available at:
https://ncbi.github.io/blast-cloud/dev/api.htmlThông thường, các đối số của hàm qblast về cơ bản tương tự với các tham số khác nhau mà bạn có thể đặt trên trang web BLAST. Điều này làm cho hàm qblast dễ hiểu cũng như giảm bớt thời gian học để sử dụng nó.
Kết nối và Tìm kiếm
Để hiểu quá trình kết nối và tìm kiếm phiên bản BLAST trực tuyến, hãy để chúng tôi thực hiện tìm kiếm theo trình tự đơn giản (có sẵn trong tệp trình tự cục bộ của chúng tôi) trên máy chủ BLAST trực tuyến thông qua Biopython.
Step 1 - Tạo một tệp có tên blast_example.fasta trong thư mục Biopython và cung cấp thông tin trình tự bên dưới làm đầu vào
Example of a single sequence in FASTA/Pearson format:
>sequence A ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattcatat
tctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtc
>sequence B ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattca
tattctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtcStep 2 - Nhập mô-đun NCBIWWW.
>>> from Bio.Blast import NCBIWWWStep 3 - Mở tệp trình tự, blast_example.fasta sử dụng mô-đun IO python.
>>> sequence_data = open("blast_example.fasta").read()
>>> sequence_data
'Example of a single sequence in FASTA/Pearson format:\n\n\n> sequence
A\nggtaagtcctctagtacaaacacccccaatattgtgatataattaaaatt
atattcatat\ntctgttgccagaaaaaacacttttaggctatattagagccatcttctttg aagcgttgtc\n\n'Step 4- Bây giờ, gọi hàm qblast truyền dữ liệu chuỗi làm tham số chính. Tham số khác đại diện cho cơ sở dữ liệu (nt) và chương trình bên trong (blastn).
>>> result_handle = NCBIWWW.qblast("blastn", "nt", sequence_data)
>>> result_handle
<_io.StringIO object at 0x000001EC9FAA4558>blast_resultsgiữ kết quả tìm kiếm của chúng tôi. Nó có thể được lưu vào một tệp để sử dụng sau này và cũng có thể được phân tích cú pháp để lấy chi tiết. Chúng ta sẽ tìm hiểu cách thực hiện trong phần tới.
Step 5 - Chức năng tương tự cũng có thể được thực hiện bằng cách sử dụng đối tượng Seq thay vì sử dụng toàn bộ tệp fasta như hình dưới đây -
>>> from Bio import SeqIO
>>> seq_record = next(SeqIO.parse(open('blast_example.fasta'),'fasta'))
>>> seq_record.id
'sequence'
>>> seq_record.seq
Seq('ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatat...gtc',
SingleLetterAlphabet())Bây giờ, hãy gọi hàm qblast truyền đối tượng Seq, record.seq làm tham số chính.
>>> result_handle = NCBIWWW.qblast("blastn", "nt", seq_record.seq)
>>> print(result_handle)
<_io.StringIO object at 0x000001EC9FAA4558>BLAST sẽ tự động chỉ định một số nhận dạng cho chuỗi của bạn.
Step 6 - Đối tượng result_handle sẽ có toàn bộ kết quả và có thể được lưu thành tệp để sử dụng sau này.
>>> with open('results.xml', 'w') as save_file:
>>> blast_results = result_handle.read()
>>> save_file.write(blast_results)Chúng ta sẽ xem cách phân tích cú pháp tệp kết quả trong phần sau.
Chạy BLAST độc lập
Phần này giải thích về cách chạy BLAST trong hệ thống cục bộ. Nếu bạn chạy BLAST trong hệ thống cục bộ, nó có thể nhanh hơn và cũng cho phép bạn tạo cơ sở dữ liệu của riêng mình để tìm kiếm theo trình tự.
Kết nối BLAST
Nói chung, chạy BLAST cục bộ không được khuyến khích do kích thước lớn, cần thêm nỗ lực để chạy phần mềm và chi phí liên quan. BLAST trực tuyến là đủ cho các mục đích cơ bản và nâng cao. Tất nhiên, đôi khi bạn có thể được yêu cầu cài đặt nó cục bộ.
Hãy xem xét bạn đang thực hiện các tìm kiếm trực tuyến thường xuyên có thể đòi hỏi nhiều thời gian và khối lượng mạng cao và nếu bạn có dữ liệu trình tự độc quyền hoặc các vấn đề liên quan đến IP, thì nên cài đặt cục bộ.
Để thực hiện việc này, chúng ta cần làm theo các bước sau:
Step 1- Tải xuống và cài đặt bản nhị phân blast mới nhất bằng liên kết đã cho - ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Step 2- Tải xuống và giải nén cơ sở dữ liệu cần thiết và mới nhất bằng liên kết dưới đây - ftp://ftp.ncbi.nlm.nih.gov/blast/db/
Phần mềm BLAST cung cấp rất nhiều cơ sở dữ liệu trong trang web của họ. Hãy để chúng tôi tải xuống tệp alu.n.gz từ trang web cơ sở dữ liệu blast và giải nén nó vào thư mục alu. Tệp này ở định dạng FASTA. Để sử dụng tệp này trong ứng dụng blast, trước tiên chúng ta cần chuyển đổi tệp từ định dạng FASTA sang định dạng cơ sở dữ liệu blast. BLAST cung cấp ứng dụng makeblastdb để thực hiện việc chuyển đổi này.
Sử dụng đoạn mã dưới đây -
cd /path/to/alu
makeblastdb -in alu.n -parse_seqids -dbtype nucl -out alunChạy đoạn mã trên sẽ phân tích cú pháp tệp đầu vào, alu.n và tạo cơ sở dữ liệu BLAST dưới dạng nhiều tệp alun.nsq, alun.nsi, v.v. Bây giờ, chúng ta có thể truy vấn cơ sở dữ liệu này để tìm chuỗi.
Chúng tôi đã cài đặt BLAST trong máy chủ cục bộ của mình và cũng có cơ sở dữ liệu BLAST mẫu, alun để truy vấn chống lại nó.
Step 3- Chúng ta hãy tạo một tệp trình tự mẫu để truy vấn cơ sở dữ liệu. Tạo một tệp search.fsa và đưa dữ liệu bên dưới vào đó.
>gnl|alu|Z15030_HSAL001056 (Alu-J)
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCT
TGAGCCTAGGAGTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAA
AGAAAAAAAAAATAGCTCTGCTGGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTG
GGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCCACGATCACACCACT
GCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
>gnl|alu|D00596_HSAL003180 (Alu-Sx)
AGCCAGGTGTGGTGGCTCACGCCTGTAATCCCACCGCTTTGGGAGGCTGAGTCAGATCAC
CTGAGGTTAGGAATTTGGGACCAGCCTGGCCAACATGGCGACACCCCAGTCTCTACTAAT
AACACAAAAAATTAGCCAGGTGTGCTGGTGCATGTCTGTAATCCCAGCTACTCAGGAGGC
TGAGGCATGAGAATTGCTCACGAGGCGGAGGTTGTAGTGAGCTGAGATCGTGGCACTGTA
CTCCAGCCTGGCGACAGAGGGAGAACCCATGTCAAAAACAAAAAAAGACACCACCAAAGG
TCAAAGCATA
>gnl|alu|X55502_HSAL000745 (Alu-J)
TGCCTTCCCCATCTGTAATTCTGGCACTTGGGGAGTCCAAGGCAGGATGATCACTTATGC
CCAAGGAATTTGAGTACCAAGCCTGGGCAATATAACAAGGCCCTGTTTCTACAAAAACTT
TAAACAATTAGCCAGGTGTGGTGGTGCGTGCCTGTGTCCAGCTACTCAGGAAGCTGAGGC
AAGAGCTTGAGGCTACAGTGAGCTGTGTTCCACCATGGTGCTCCAGCCTGGGTGACAGGG
CAAGACCCTGTCAAAAGAAAGGAAGAAAGAACGGAAGGAAAGAAGGAAAGAAACAAGGAG
AGDữ liệu trình tự được thu thập từ tệp alu.n; do đó, nó phù hợp với cơ sở dữ liệu của chúng tôi.
Step 4 - Phần mềm BLAST cung cấp nhiều ứng dụng để tìm kiếm cơ sở dữ liệu và chúng tôi sử dụng blastn. blastn application requires minimum of three arguments, db, query and out. db đề cập đến cơ sở dữ liệu chống lại để tìm kiếm; query là trình tự để khớp và outlà tệp để lưu trữ kết quả. Bây giờ, hãy chạy lệnh dưới đây để thực hiện truy vấn đơn giản này -
blastn -db alun -query search.fsa -out results.xml -outfmt 5Chạy lệnh trên sẽ tìm kiếm và đưa ra kết quả trong results.xml tệp như được cung cấp bên dưới (một phần dữ liệu) -
<?xml version = "1.0"?>
<!DOCTYPE BlastOutput PUBLIC "-//NCBI//NCBI BlastOutput/EN"
"http://www.ncbi.nlm.nih.gov/dtd/NCBI_BlastOutput.dtd">
<BlastOutput>
<BlastOutput_program>blastn</BlastOutput_program>
<BlastOutput_version>BLASTN 2.7.1+</BlastOutput_version>
<BlastOutput_reference>Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
</BlastOutput_reference>
<BlastOutput_db>alun</BlastOutput_db>
<BlastOutput_query-ID>Query_1</BlastOutput_query-ID>
<BlastOutput_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</BlastOutput_query-def>
<BlastOutput_query-len>292</BlastOutput_query-len>
<BlastOutput_param>
<Parameters>
<Parameters_expect>10</Parameters_expect>
<Parameters_sc-match>1</Parameters_sc-match>
<Parameters_sc-mismatch>-2</Parameters_sc-mismatch>
<Parameters_gap-open>0</Parameters_gap-open>
<Parameters_gap-extend>0</Parameters_gap-extend>
<Parameters_filter>L;m;</Parameters_filter>
</Parameters>
</BlastOutput_param>
<BlastOutput_iterations>
<Iteration>
<Iteration_iter-num>1</Iteration_iter-num><Iteration_query-ID>Query_1</Iteration_query-ID>
<Iteration_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</Iteration_query-def>
<Iteration_query-len>292</Iteration_query-len>
<Iteration_hits>
<Hit>
<Hit_num>1</Hit_num>
<Hit_id>gnl|alu|Z15030_HSAL001056</Hit_id>
<Hit_def>(Alu-J)</Hit_def>
<Hit_accession>Z15030_HSAL001056</Hit_accession>
<Hit_len>292</Hit_len>
<Hit_hsps>
<Hsp>
<Hsp_num>1</Hsp_num>
<Hsp_bit-score>540.342</Hsp_bit-score>
<Hsp_score>292</Hsp_score>
<Hsp_evalue>4.55414e-156</Hsp_evalue>
<Hsp_query-from>1</Hsp_query-from>
<Hsp_query-to>292</Hsp_query-to>
<Hsp_hit-from>1</Hsp_hit-from>
<Hsp_hit-to>292</Hsp_hit-to>
<Hsp_query-frame>1</Hsp_query-frame>
<Hsp_hit-frame>1</Hsp_hit-frame>
<Hsp_identity>292</Hsp_identity>
<Hsp_positive>292</Hsp_positive>
<Hsp_gaps>0</Hsp_gaps>
<Hsp_align-len>292</Hsp_align-len>
<Hsp_qseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGAGTTTG
CGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCTGGTGGTGCATG
CCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCC
ACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
</Hsp_qseq>
<Hsp_hseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGA
GTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCT
GGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGG
CTGTGGTGAGCCACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAAC
AAATAA
</Hsp_hseq>
<Hsp_midline>
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||
</Hsp_midline>
</Hsp>
</Hit_hsps>
</Hit>
.........................
.........................
.........................
</Iteration_hits>
<Iteration_stat>
<Statistics>
<Statistics_db-num>327</Statistics_db-num>
<Statistics_db-len>80506</Statistics_db-len>
<Statistics_hsp-lenv16</Statistics_hsp-len>
<Statistics_eff-space>21528364</Statistics_eff-space>
<Statistics_kappa>0.46</Statistics_kappa>
<Statistics_lambda>1.28</Statistics_lambda>
<Statistics_entropy>0.85</Statistics_entropy>
</Statistics>
</Iteration_stat>
</Iteration>
</BlastOutput_iterations>
</BlastOutput>Lệnh trên có thể được chạy bên trong python bằng đoạn mã dưới đây:
>>> from Bio.Blast.Applications import NcbiblastnCommandline
>>> blastn_cline = NcbiblastnCommandline(query = "search.fasta", db = "alun",
outfmt = 5, out = "results.xml")
>>> stdout, stderr = blastn_cline()Ở đây, cái đầu tiên là một xử lý cho đầu ra blast và cái thứ hai là đầu ra lỗi có thể được tạo ra bởi lệnh blast.
Vì chúng tôi đã cung cấp tệp đầu ra dưới dạng đối số dòng lệnh (out = “results.xml”) và đặt định dạng đầu ra là XML (outfmt = 5), tệp đầu ra sẽ được lưu trong thư mục làm việc hiện tại.
Phân tích cú pháp kết quả BLAST
Nói chung, đầu ra BLAST được phân tích cú pháp dưới dạng định dạng XML bằng cách sử dụng mô-đun NCBIXML. Để thực hiện việc này, chúng ta cần nhập mô-đun sau:
>>> from Bio.Blast import NCBIXMLHiện nay, open the file directly using python open method và use NCBIXML parse method như dưới đây -
>>> E_VALUE_THRESH = 1e-20
>>> for record in NCBIXML.parse(open("results.xml")):
>>> if record.alignments:
>>> print("\n")
>>> print("query: %s" % record.query[:100])
>>> for align in record.alignments:
>>> for hsp in align.hsps:
>>> if hsp.expect < E_VALUE_THRESH:
>>> print("match: %s " % align.title[:100])Điều này sẽ tạo ra một đầu ra như sau:
query: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|L12964_HSAL003860 (Alu-J)
match: gnl|alu|L13042_HSAL003863 (Alu-FLA?)
match: gnl|alu|M86249_HSAL001462 (Alu-FLA?)
match: gnl|alu|M29484_HSAL002265 (Alu-J)
query: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|J03071_HSAL001860 (Alu-J)
match: gnl|alu|X72409_HSAL005025 (Alu-Sx)
query: gnl|alu|X55502_HSAL000745 (Alu-J)
match: gnl|alu|X55502_HSAL000745 (Alu-J)Entrezlà một hệ thống tìm kiếm trực tuyến do NCBI cung cấp. Nó cung cấp quyền truy cập vào gần như tất cả các cơ sở dữ liệu sinh học phân tử đã biết với một truy vấn toàn cầu tích hợp hỗ trợ các toán tử Boolean và tìm kiếm thực địa. Nó trả về kết quả từ tất cả cơ sở dữ liệu với thông tin như số lần truy cập từ mỗi cơ sở dữ liệu, bản ghi có liên kết đến cơ sở dữ liệu gốc, v.v.
Một số cơ sở dữ liệu phổ biến có thể được truy cập thông qua Entrez được liệt kê dưới đây:
- Pubmed
- Pubmed Central
- Nucleotide (Cơ sở dữ liệu trình tự GenBank)
- Protein (Cơ sở dữ liệu trình tự)
- Bộ gen (Toàn bộ cơ sở dữ liệu bộ gen)
- Cấu trúc (Cấu trúc đại phân tử ba chiều)
- Phân loại học (Sinh vật trong Ngân hàng gen)
- SNP (Đa hình Nucleotide Đơn)
- UniGene (Cụm chuỗi phiên mã được định hướng chung)
- CDD (Cơ sở dữ liệu miền protein được bảo tồn)
- Miền 3D (Miền từ cấu trúc Entrez)
Ngoài các cơ sở dữ liệu trên, Entrez còn cung cấp nhiều cơ sở dữ liệu khác để thực hiện việc tìm kiếm thực địa.
Biopython cung cấp một mô-đun cụ thể của Entrez, Bio.Entrez để truy cập cơ sở dữ liệu Entrez. Hãy để chúng tôi tìm hiểu cách truy cập Entrez bằng Biopython trong chương này -
Các bước kết nối cơ sở dữ liệu
Để thêm các tính năng của Entrez, hãy nhập mô-đun sau:
>>> from Bio import EntrezTiếp theo, đặt email của bạn để xác định ai được kết nối với mã được cung cấp bên dưới -
>>> Entrez.email = '<youremail>'Sau đó, đặt thông số công cụ Entrez và theo mặc định, nó là Biopython.
>>> Entrez.tool = 'Demoscript'Hiện nay, call einfo function to find index term counts, last update, and available links for each database như được định nghĩa bên dưới -
>>> info = Entrez.einfo()Phương thức einfo trả về một đối tượng, đối tượng này cung cấp quyền truy cập vào thông tin thông qua phương thức đọc của nó như được hiển thị bên dưới:
>>> data = info.read()
>>> print(data)
<?xml version = "1.0" encoding = "UTF-8" ?>
<!DOCTYPE eInfoResult PUBLIC "-//NLM//DTD einfo 20130322//EN"
"https://eutils.ncbi.nlm.nih.gov/eutils/dtd/20130322/einfo.dtd">
<eInfoResult>
<DbList>
<DbName>pubmed</DbName>
<DbName>protein</DbName>
<DbName>nuccore</DbName>
<DbName>ipg</DbName>
<DbName>nucleotide</DbName>
<DbName>nucgss</DbName>
<DbName>nucest</DbName>
<DbName>structure</DbName>
<DbName>sparcle</DbName>
<DbName>genome</DbName>
<DbName>annotinfo</DbName>
<DbName>assembly</DbName>
<DbName>bioproject</DbName>
<DbName>biosample</DbName>
<DbName>blastdbinfo</DbName>
<DbName>books</DbName>
<DbName>cdd</DbName>
<DbName>clinvar</DbName>
<DbName>clone</DbName>
<DbName>gap</DbName>
<DbName>gapplus</DbName>
<DbName>grasp</DbName>
<DbName>dbvar</DbName>
<DbName>gene</DbName>
<DbName>gds</DbName>
<DbName>geoprofiles</DbName>
<DbName>homologene</DbName>
<DbName>medgen</DbName>
<DbName>mesh</DbName>
<DbName>ncbisearch</DbName>
<DbName>nlmcatalog</DbName>
<DbName>omim</DbName>
<DbName>orgtrack</DbName>
<DbName>pmc</DbName>
<DbName>popset</DbName>
<DbName>probe</DbName>
<DbName>proteinclusters</DbName>
<DbName>pcassay</DbName>
<DbName>biosystems</DbName>
<DbName>pccompound</DbName>
<DbName>pcsubstance</DbName>
<DbName>pubmedhealth</DbName>
<DbName>seqannot</DbName>
<DbName>snp</DbName>
<DbName>sra</DbName>
<DbName>taxonomy</DbName>
<DbName>biocollections</DbName>
<DbName>unigene</DbName>
<DbName>gencoll</DbName>
<DbName>gtr</DbName>
</DbList>
</eInfoResult>Dữ liệu ở định dạng XML và để lấy dữ liệu dưới dạng đối tượng python, hãy sử dụng Entrez.read phương pháp càng sớm càng tốt Entrez.einfo() phương thức được gọi -
>>> info = Entrez.einfo()
>>> record = Entrez.read(info)Ở đây, bản ghi là một từ điển có một khóa, DbList như hình dưới đây -
>>> record.keys()
[u'DbList']Truy cập vào khóa DbList trả về danh sách tên cơ sở dữ liệu được hiển thị bên dưới:
>>> record[u'DbList']
['pubmed', 'protein', 'nuccore', 'ipg', 'nucleotide', 'nucgss',
'nucest', 'structure', 'sparcle', 'genome', 'annotinfo', 'assembly',
'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar',
'clone', 'gap', 'gapplus', 'grasp', 'dbvar', 'gene', 'gds', 'geoprofiles',
'homologene', 'medgen', 'mesh', 'ncbisearch', 'nlmcatalog', 'omim',
'orgtrack', 'pmc', 'popset', 'probe', 'proteinclusters', 'pcassay',
'biosystems', 'pccompound', 'pcsubstance', 'pubmedhealth', 'seqannot',
'snp', 'sra', 'taxonomy', 'biocollections', 'unigene', 'gencoll', 'gtr']
>>>Về cơ bản, mô-đun Entrez phân tích cú pháp XML do hệ thống tìm kiếm Entrez trả về và cung cấp nó dưới dạng danh sách và từ điển python.
Cơ sở dữ liệu tìm kiếm
Để tìm kiếm bất kỳ một trong các cơ sở dữ liệu Entrez, chúng ta có thể sử dụng mô-đun Bio.Entrez.esearch (). Nó được định nghĩa dưới đây -
>>> info = Entrez.einfo()
>>> info = Entrez.esearch(db = "pubmed",term = "genome")
>>> record = Entrez.read(info)
>>>print(record)
DictElement({u'Count': '1146113', u'RetMax': '20', u'IdList':
['30347444', '30347404', '30347317', '30347292',
'30347286', '30347249', '30347194', '30347187',
'30347172', '30347088', '30347075', '30346992',
'30346990', '30346982', '30346980', '30346969',
'30346962', '30346954', '30346941', '30346939'],
u'TranslationStack': [DictElement({u'Count':
'927819', u'Field': 'MeSH Terms', u'Term': '"genome"[MeSH Terms]',
u'Explode': 'Y'}, attributes = {})
, DictElement({u'Count': '422712', u'Field':
'All Fields', u'Term': '"genome"[All Fields]', u'Explode': 'N'}, attributes = {}),
'OR', 'GROUP'], u'TranslationSet': [DictElement({u'To': '"genome"[MeSH Terms]
OR "genome"[All Fields]', u'From': 'genome'}, attributes = {})], u'RetStart': '0',
u'QueryTranslation': '"genome"[MeSH Terms] OR "genome"[All Fields]'},
attributes = {})
>>>Nếu bạn gán db không chính xác thì nó sẽ trả về
>>> info = Entrez.esearch(db = "blastdbinfo",term = "books")
>>> record = Entrez.read(info)
>>> print(record)
DictElement({u'Count': '0', u'RetMax': '0', u'IdList': [],
u'WarningList': DictElement({u'OutputMessage': ['No items found.'],
u'PhraseIgnored': [], u'QuotedPhraseNotFound': []}, attributes = {}),
u'ErrorList': DictElement({u'FieldNotFound': [], u'PhraseNotFound':
['books']}, attributes = {}), u'TranslationSet': [], u'RetStart': '0',
u'QueryTranslation': '(books[All Fields])'}, attributes = {})Nếu bạn muốn tìm kiếm trên cơ sở dữ liệu, thì bạn có thể sử dụng Entrez.egquery. Điều này tương tự nhưEntrez.esearch ngoại trừ nó là đủ để chỉ định từ khóa và bỏ qua tham số cơ sở dữ liệu.
>>>info = Entrez.egquery(term = "entrez")
>>> record = Entrez.read(info)
>>> for row in record["eGQueryResult"]:
... print(row["DbName"], row["Count"])
...
pubmed 458
pmc 12779 mesh 1
...
...
...
biosample 7
biocollections 0Tìm nạp bản ghi
Enterz cung cấp một phương pháp đặc biệt, efetch để tìm kiếm và tải xuống toàn bộ chi tiết của một bản ghi từ Entrez. Hãy xem xét ví dụ đơn giản sau:
>>> handle = Entrez.efetch(
db = "nucleotide", id = "EU490707", rettype = "fasta")Bây giờ, chúng ta có thể chỉ cần đọc các bản ghi bằng cách sử dụng đối tượng SeqIO
>>> record = SeqIO.read( handle, "fasta" )
>>> record
SeqRecord(seq = Seq('ATTTTTTACGAACCTGTGGAAATTTTTGGTTATGACAATAAATCTAGTTTAGTA...GAA',
SingleLetterAlphabet()), id = 'EU490707.1', name = 'EU490707.1',
description = 'EU490707.1
Selenipedium aequinoctiale maturase K (matK) gene, partial cds; chloroplast',
dbxrefs = [])Biopython cung cấp mô-đun Bio.PDB để thao tác các cấu trúc polypeptide. PDB (Ngân hàng Dữ liệu Protein) là nguồn tài nguyên cấu trúc protein lớn nhất hiện có trên mạng. Nó chứa rất nhiều cấu trúc protein riêng biệt, bao gồm phức hợp protein-protein, protein-DNA, protein-RNA.
Để tải PDB, hãy nhập lệnh dưới đây:
from Bio.PDB import *Định dạng tệp cấu trúc protein
PDB phân phối cấu trúc protein ở ba định dạng khác nhau -
- Định dạng tệp dựa trên XML không được Biopython hỗ trợ
- Định dạng tệp pdb, là tệp văn bản được định dạng đặc biệt
- Định dạng tệp PDBx / mmCIF
Các tệp PDB do Ngân hàng Dữ liệu Protein phân phối có thể chứa các lỗi định dạng khiến chúng trở nên mơ hồ hoặc khó phân tích cú pháp. Mô-đun Bio.PDB cố gắng tự động xử lý các lỗi này.
Mô-đun Bio.PDB triển khai hai trình phân tích cú pháp khác nhau, một là định dạng mmCIF và một là định dạng pdb.
Hãy để chúng tôi tìm hiểu cách phân tích cú pháp chi tiết từng định dạng -
Bộ phân tích cú pháp mmCIF
Hãy để chúng tôi tải xuống cơ sở dữ liệu mẫu ở định dạng mmCIF từ máy chủ pdb bằng lệnh dưới đây:
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'mmCif')Thao tác này sẽ tải xuống tệp được chỉ định (2fat.cif) từ máy chủ và lưu trữ trong thư mục làm việc hiện tại.
Tại đây, PDBList cung cấp các tùy chọn để liệt kê và tải xuống các tệp từ máy chủ PDB FTP trực tuyến. Phương thức get_pdb_file cần tên của tệp được tải xuống mà không có phần mở rộng. get_pdb_file cũng có tùy chọn để chỉ định thư mục tải xuống, pdir và định dạng của tệp, file_format. Các giá trị có thể có của định dạng tệp như sau:
- “MmCif” (mặc định, tệp PDBx / mmCif)
- “Pdb” (định dạng PDB)
- “Xml” (định dạng PMDML / XML)
- “Mmtf” (nén cao)
- “Gói” (kho lưu trữ định dạng PDB cho cấu trúc lớn)
Để tải tệp cif, hãy sử dụng Bio.MMCIF.MMCIFParser như được chỉ định bên dưới -
>>> parser = MMCIFParser(QUIET = True)
>>> data = parser.get_structure("2FAT", "2FAT.cif")Ở đây, QUIET ngăn chặn cảnh báo trong quá trình phân tích cú pháp tệp. get_structure will parse the file and return the structure with id as 2FAT (đối số đầu tiên).
Sau khi chạy lệnh trên, nó phân tích cú pháp tệp và in cảnh báo có thể có, nếu có.
Bây giờ, hãy kiểm tra cấu trúc bằng lệnh dưới đây:
>>> data
<Structure id = 2FAT>
To get the type, use type method as specified below,
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Chúng tôi đã phân tích cú pháp thành công tệp và có cấu trúc của protein. Chúng ta sẽ tìm hiểu chi tiết về cấu trúc của protein và cách lấy nó trong chương sau.
PDB Parser
Hãy để chúng tôi tải xuống cơ sở dữ liệu mẫu ở định dạng PDB từ máy chủ pdb bằng lệnh dưới đây:
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'pdb')Thao tác này sẽ tải xuống tệp được chỉ định (pdb2fat.ent) từ máy chủ và lưu trữ trong thư mục làm việc hiện tại.
Để tải tệp pdb, hãy sử dụng Bio.PDB.PDBParser như được chỉ định bên dưới -
>>> parser = PDBParser(PERMISSIVE = True, QUIET = True)
>>> data = parser.get_structure("2fat","pdb2fat.ent")Ở đây, get_ architecture tương tự như MMCIFParser. Tùy chọn PERMISSIVE cố gắng phân tích cú pháp dữ liệu protein linh hoạt nhất có thể.
Bây giờ, hãy kiểm tra cấu trúc và loại của nó với đoạn mã được cung cấp bên dưới -
>>> data
<Structure id = 2fat>
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Vâng, cấu trúc tiêu đề lưu trữ thông tin từ điển. Để thực hiện việc này, hãy nhập lệnh dưới đây:
>>> print(data.header.keys()) dict_keys([
'name', 'head', 'deposition_date', 'release_date', 'structure_method', 'resolution',
'structure_reference', 'journal_reference', 'author', 'compound', 'source',
'keywords', 'journal'])
>>>Để lấy tên, hãy sử dụng mã sau:
>>> print(data.header["name"])
an anti-urokinase plasminogen activator receptor (upar) antibody: crystal
structure and binding epitope
>>>Bạn cũng có thể kiểm tra ngày tháng và độ phân giải bằng mã bên dưới -
>>> print(data.header["release_date"]) 2006-11-14
>>> print(data.header["resolution"]) 1.77Cấu trúc PDB
Cấu trúc PDB bao gồm một mô hình duy nhất, chứa hai chuỗi.
- chuỗi L, chứa số lượng dư
- chuỗi H, chứa số dư
Mỗi phần dư bao gồm nhiều nguyên tử, mỗi nguyên tử có một vị trí 3D được biểu thị bằng tọa độ (x, y, z).
Hãy cùng chúng tôi tìm hiểu cách nhận cấu trúc của nguyên tử một cách chi tiết trong phần dưới đây -
Mô hình
Phương thức Structure.get_models () trả về một trình lặp trên các mô hình. Nó được định nghĩa dưới đây -
>>> model = data.get_models()
>>> model
<generator object get_models at 0x103fa1c80>
>>> models = list(model)
>>> models [<Model id = 0>]
>>> type(models[0])
<class 'Bio.PDB.Model.Model'>Ở đây, Mô hình mô tả chính xác một cấu trúc 3D. Nó chứa một hoặc nhiều chuỗi.
Chuỗi
Phương thức Model.get_chain () trả về một trình vòng lặp trên các chuỗi. Nó được định nghĩa dưới đây -
>>> chains = list(models[0].get_chains())
>>> chains
[<Chain id = L>, <Chain id = H>]
>>> type(chains[0])
<class 'Bio.PDB.Chain.Chain'>Ở đây, Chuỗi mô tả một cấu trúc polypeptit thích hợp, tức là một chuỗi các gốc liên kết liên tiếp.
Phần còn lại
Phương thức Chain.get_residues () trả về một trình lặp trên các phần còn lại. Nó được định nghĩa dưới đây -
>>> residue = list(chains[0].get_residues())
>>> len(residue)
293
>>> residue1 = list(chains[1].get_residues())
>>> len(residue1)
311Chà, Dư lượng giữ các nguyên tử thuộc về một axit amin.
Nguyên tử
Residue.get_atom () trả về một trình lặp trên các nguyên tử như được định nghĩa bên dưới:
>>> atoms = list(residue[0].get_atoms())
>>> atoms
[<Atom N>, <Atom CA>, <Atom C>, <Atom Ov, <Atom CB>, <Atom CG>, <Atom OD1>, <Atom OD2>]Một nguyên tử giữ tọa độ 3D của một nguyên tử và nó được gọi là Vector. Nó được định nghĩa dưới đây
>>> atoms[0].get_vector()
<Vector 18.49, 73.26, 44.16>Nó đại diện cho các giá trị tọa độ x, y và z.
Mô típ trình tự là mô hình trình tự nucleotit hoặc axit amin. Các mô típ trình tự được hình thành bởi sự sắp xếp ba chiều của các axit amin có thể không liền nhau. Biopython cung cấp một mô-đun riêng biệt, Bio.motifs để truy cập các chức năng của mô típ trình tự như được chỉ định bên dưới:
from Bio import motifsTạo mô hình DNA đơn giản
Hãy để chúng tôi tạo một chuỗi mô típ DNA đơn giản bằng lệnh dưới đây:
>>> from Bio import motifs
>>> from Bio.Seq import Seq
>>> DNA_motif = [ Seq("AGCT"),
... Seq("TCGA"),
... Seq("AACT"),
... ]
>>> seq = motifs.create(DNA_motif)
>>> print(seq) AGCT TCGA AACTĐể đếm các giá trị chuỗi, hãy sử dụng lệnh dưới đây:
>>> print(seq.counts)
0 1 2 3
A: 2.00 1.00 0.00 1.00
C: 0.00 1.00 2.00 0.00
G: 0.00 1.00 1.00 0.00
T: 1.00 0.00 0.00 2.00Sử dụng mã sau để đếm 'A' trong chuỗi:
>>> seq.counts["A", :]
(2, 1, 0, 1)Nếu bạn muốn truy cập các cột đếm, hãy sử dụng lệnh dưới đây:
>>> seq.counts[:, 3]
{'A': 1, 'C': 0, 'T': 2, 'G': 0}Tạo biểu trưng trình tự
Bây giờ chúng ta sẽ thảo luận về cách tạo Biểu trưng trình tự.
Hãy xem xét trình tự dưới đây -
AGCTTACG
ATCGTACC
TTCCGAAT
GGTACGTA
AAGCTTGGBạn có thể tạo biểu trưng của riêng mình bằng liên kết sau: http://weblogo.berkeley.edu/
Thêm trình tự trên và tạo một logo mới và lưu hình ảnh có tên seq.png trong thư mục biopython của bạn.
seq.pngSau khi tạo hình ảnh, bây giờ hãy chạy lệnh sau:
>>> seq.weblogo("seq.png")Mô típ trình tự DNA này được thể hiện dưới dạng biểu trưng trình tự cho mô típ liên kết LexA.
Cơ sở dữ liệu JASPAR
JASPAR là một trong những cơ sở dữ liệu phổ biến nhất. Nó cung cấp các phương tiện của bất kỳ định dạng motif nào để đọc, viết và quét trình tự. Nó lưu trữ siêu thông tin cho từng mô típ.The module Bio.motifs contains a specialized class jaspar.Motif to represent meta-information attributes.
Nó có các loại thuộc tính đáng chú ý sau:
- matrix_id - ID mô típ JASPAR duy nhất
- tên - Tên của mô típ
- tf_family - Họ mô-típ, ví dụ: 'Helix-Loop-Helix'
- data_type - kiểu dữ liệu được sử dụng trong motif.
Hãy để chúng tôi tạo một định dạng trang web JASPAR có tên trong sample.sites trong thư mục biopython. Nó được định nghĩa dưới đây -
sample.sites
>MA0001 ARNT 1
AACGTGatgtccta
>MA0001 ARNT 2
CAGGTGggatgtac
>MA0001 ARNT 3
TACGTAgctcatgc
>MA0001 ARNT 4
AACGTGacagcgct
>MA0001 ARNT 5
CACGTGcacgtcgt
>MA0001 ARNT 6
cggcctCGCGTGcTrong tệp trên, chúng tôi đã tạo các thể hiện mô típ. Bây giờ, chúng ta hãy tạo một đối tượng motif từ các trường hợp trên -
>>> from Bio import motifs
>>> with open("sample.sites") as handle:
... data = motifs.read(handle,"sites")
...
>>> print(data)
TF name None
Matrix ID None
Matrix:
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00Tại đây, dữ liệu đọc tất cả các phiên bản motif từ tệp sample.sites.
Để in tất cả các phiên bản từ dữ liệu, hãy sử dụng lệnh dưới đây:
>>> for instance in data.instances:
... print(instance)
...
AACGTG
CAGGTG
TACGTA
AACGTG
CACGTG
CGCGTGSử dụng lệnh dưới đây để đếm tất cả các giá trị -
>>> print(data.counts)
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00
>>>BioSQLlà một lược đồ cơ sở dữ liệu chung được thiết kế chủ yếu để lưu trữ trình tự và dữ liệu liên quan của nó cho tất cả công cụ RDBMS. Nó được thiết kế theo cách giữ dữ liệu từ tất cả các cơ sở dữ liệu tin sinh học phổ biến như GenBank, Swissport, v.v. Nó cũng có thể được sử dụng để lưu trữ dữ liệu nội bộ.
BioSQL hiện đang cung cấp lược đồ cụ thể cho các cơ sở dữ liệu bên dưới -
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Nó cũng cung cấp hỗ trợ tối thiểu cho cơ sở dữ liệu HSQLDB và Derby dựa trên Java.
BioPython cung cấp khả năng ORM rất đơn giản, dễ dàng và nâng cao để làm việc với cơ sở dữ liệu dựa trên BioSQL. BioPython provides a module, BioSQL để thực hiện chức năng sau -
- Tạo / xóa cơ sở dữ liệu BioSQL
- Kết nối với cơ sở dữ liệu BioSQL
- Phân tích cú pháp một cơ sở dữ liệu trình tự như GenBank, Swisport, kết quả BLAST, kết quả Entrez, v.v. và tải trực tiếp nó vào cơ sở dữ liệu BioSQL
- Tìm nạp dữ liệu trình tự từ cơ sở dữ liệu BioSQL
- Tìm nạp dữ liệu phân loại từ NCBI BLAST và lưu trữ trong cơ sở dữ liệu BioSQL
- Chạy bất kỳ truy vấn SQL nào đối với cơ sở dữ liệu BioSQL
Tổng quan về lược đồ cơ sở dữ liệu BioSQL
Trước khi đi sâu vào BioSQL, chúng ta hãy hiểu cơ bản về lược đồ BioSQL. Lược đồ BioSQL cung cấp hơn 25 bảng để chứa dữ liệu trình tự, tính năng trình tự, danh mục trình tự / bản thể học và thông tin phân loại. Một số bảng quan trọng như sau:
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
Tạo cơ sở dữ liệu BioSQL
Trong phần này, chúng ta hãy tạo cơ sở dữ liệu BioSQL mẫu, biosql bằng cách sử dụng lược đồ do nhóm BioSQL cung cấp. Chúng tôi sẽ làm việc với cơ sở dữ liệu SQLite vì nó thực sự dễ bắt đầu và không phải thiết lập phức tạp.
Ở đây, chúng tôi sẽ tạo cơ sở dữ liệu BioSQL dựa trên SQLite bằng cách sử dụng các bước dưới đây.
Step 1 - Tải xuống công cụ dữ liệu SQLite và cài đặt nó.
Step 2 - Tải xuống dự án BioSQL từ URL GitHub. https://github.com/biosql/biosql
Step 3 - Mở bảng điều khiển và tạo thư mục bằng mkdir và nhập vào đó.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 - Chạy lệnh dưới đây để tạo cơ sở dữ liệu SQLite mới.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 - Sao chép tệp biosqldb-sqlite.sql từ dự án BioSQL (/ sql / biosqldb-sqlite.sql`) và lưu trữ trong thư mục hiện tại.
Step 6 - Chạy lệnh dưới đây để tạo tất cả các bảng.
sqlite> .read biosqldb-sqlite.sqlBây giờ, tất cả các bảng được tạo trong cơ sở dữ liệu mới của chúng tôi.
Step 7 - Chạy lệnh dưới đây để xem tất cả các bảng mới trong cơ sở dữ liệu của chúng tôi.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>Ba lệnh đầu tiên là các lệnh cấu hình để cấu hình SQLite hiển thị kết quả theo cách đã định dạng.
Step 8 - Sao chép tệp GenBank mẫu, ls_orchid.gbk do nhóm BioPython cung cấp https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk vào thư mục hiện tại và lưu nó dưới dạng lan.gbk.
Step 9 - Tạo một tập lệnh python, load_orchid.py bằng cách sử dụng đoạn mã dưới đây và thực thi nó.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Đoạn mã trên phân tích cú pháp bản ghi trong tệp và chuyển đổi nó thành các đối tượng python và chèn nó vào cơ sở dữ liệu BioSQL. Chúng tôi sẽ phân tích mã trong phần sau.
Cuối cùng, chúng tôi đã tạo một cơ sở dữ liệu BioSQL mới và tải một số dữ liệu mẫu vào đó. Chúng ta sẽ thảo luận về các bảng quan trọng trong chương tiếp theo.
Sơ đồ ER đơn giản
biodatabase bảng nằm trên cùng của hệ thống phân cấp và mục đích chính của nó là tổ chức một tập hợp dữ liệu trình tự thành một nhóm / cơ sở dữ liệu ảo. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. Tất cả các bảng liên quan trong cơ sở dữ liệu BioSQL đều có tham chiếu đến mục nhập cơ sở dữ liệu sinh học.
bioentrybảng chứa tất cả các chi tiết về một chuỗi ngoại trừ dữ liệu chuỗi. dữ liệu trình tự của một cụ thểbioentry sẽ được lưu trữ trong biosequence bàn.
taxon và taxon_name là các chi tiết phân loại và mọi mục nhập đều tham chiếu đến bảng này để chỉ định thông tin về đơn vị phân loại của nó.
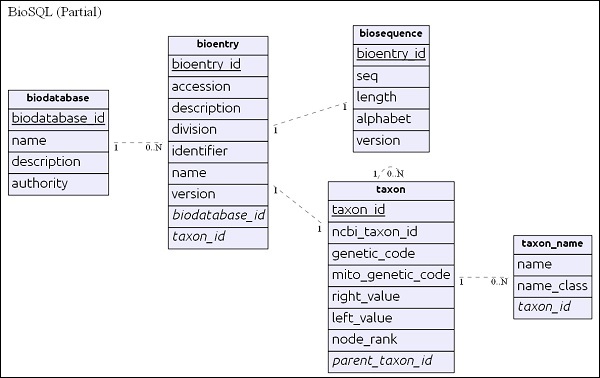
Sau khi hiểu lược đồ, chúng ta hãy xem xét một số truy vấn trong phần tiếp theo.
Truy vấn BioSQL
Chúng ta hãy đi sâu vào một số truy vấn SQL để hiểu rõ hơn cách dữ liệu được tổ chức và các bảng có liên quan với nhau. Trước khi tiếp tục, chúng ta hãy mở cơ sở dữ liệu bằng lệnh dưới đây và đặt một số lệnh định dạng -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Bạn cũng có thể sử dụng bất kỳ trình soạn thảo SQLite nào để chạy truy vấn.
Liệt kê cơ sở dữ liệu chuỗi ảo có sẵn trong hệ thống như dưới đây:
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Ở đây, chúng tôi chỉ có một cơ sở dữ liệu, orchid.
Liệt kê các mục nhập (đầu trang 3) có sẵn trong cơ sở dữ liệu orchid với mã cho sẵn dưới đây
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Liệt kê chi tiết trình tự được liên kết với mục nhập (gia nhập - Z78530, tên - gen C. fasciculatum 5.8S rRNA và DNA ITS1 và ITS2) với mã đã cho -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Nhận trình tự hoàn chỉnh được liên kết với mục nhập (gia nhập - Z78530, tên - gen C. fasciculatum 5.8S rRNA và DNA ITS1 và ITS2) bằng cách sử dụng mã dưới đây -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Liệt kê đơn vị phân loại liên kết với cơ sở dữ liệu sinh học, phong lan
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>Tải dữ liệu vào cơ sở dữ liệu BioSQL
Hãy để chúng tôi tìm hiểu cách tải dữ liệu tuần tự vào cơ sở dữ liệu BioSQL trong chương này. Chúng tôi đã có mã để tải dữ liệu vào cơ sở dữ liệu trong phần trước và mã như sau:
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Chúng ta sẽ có cái nhìn sâu hơn về từng dòng mã và mục đích của nó -
Line 1 - Tải mô-đun SeqIO.
Line 2- Tải mô-đun BioSeqDatabase. Mô-đun này cung cấp tất cả các chức năng để tương tác với cơ sở dữ liệu BioSQL.
Line 3 - Tải mô-đun os.
Line 5- open_database mở cơ sở dữ liệu được chỉ định (db) với trình điều khiển (trình điều khiển) được cấu hình và trả về một xử lý cho cơ sở dữ liệu BioSQL (máy chủ). Biopython hỗ trợ cơ sở dữ liệu sqlite, mysql, postgresql và oracle.
Line 6-10- Phương thức load_database_sql tải sql từ tệp bên ngoài và thực thi nó. phương thức cam kết thực hiện giao dịch. Chúng ta có thể bỏ qua bước này vì chúng ta đã tạo cơ sở dữ liệu bằng lược đồ.
Line 12 - Các phương thức new_database tạo cơ sở dữ liệu ảo mới, lan và trả về một db xử lý để thực thi lệnh đối với cơ sở dữ liệu lan.
Line 13- load method tải các mục trình tự (SeqRecord có thể lặp lại) vào cơ sở dữ liệu lan. SqlIO.parse phân tích cú pháp cơ sở dữ liệu GenBank và trả về tất cả các chuỗi trong đó dưới dạng SeqRecord có thể lặp lại. Tham số thứ hai (True) của phương thức tải hướng dẫn nó tìm nạp các chi tiết phân loại của dữ liệu trình tự từ trang web nổ NCBI, nếu nó chưa có sẵn trong hệ thống.
Line 14 - cam kết cam kết giao dịch.
Line 15 - Đóng đóng kết nối cơ sở dữ liệu và phá hủy xử lý máy chủ.
Tìm nạp dữ liệu trình tự
Hãy để chúng tôi tìm nạp một chuỗi với số nhận dạng, 2765658 từ cơ sở dữ liệu phong lan như bên dưới -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Ở đây, máy chủ ["Orchid"] trả về xử lý để tìm nạp dữ liệu từ cơ sở dữ liệu ảo. lookup phương pháp cung cấp một tùy chọn để chọn trình tự dựa trên tiêu chí và chúng tôi đã chọn trình tự có số nhận dạng, 2765658. lookuptrả về thông tin trình tự dưới dạng SeqRecordobject. Vì chúng tôi đã biết cách làm việc với SeqRecord`, nên việc lấy dữ liệu từ nó rất dễ dàng.
Xóa cơ sở dữ liệu
Việc xóa cơ sở dữ liệu cũng đơn giản như gọi phương thức remove_database với tên cơ sở dữ liệu thích hợp và sau đó cam kết nó như được chỉ định bên dưới:
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()Di truyền quần thể đóng một vai trò quan trọng trong thuyết tiến hóa. Nó phân tích sự khác biệt di truyền giữa các loài cũng như hai hoặc nhiều cá thể trong cùng một loài.
Biopython cung cấp mô-đun Bio.PopGen cho di truyền quần thể và chủ yếu hỗ trợ `GenePop, một gói di truyền phổ biến được phát triển bởi Michel Raymond và Francois Rousset.
Một trình phân tích cú pháp đơn giản
Hãy để chúng tôi viết một ứng dụng đơn giản để phân tích định dạng GenePop và hiểu khái niệm này.
Tải xuống tệp genPop do nhóm Biopython cung cấp theo liên kết dưới đây -https://raw.githubusercontent.com/biopython/biopython/master/Tests/PopGen/c3line.gen
Tải mô-đun GenePop bằng đoạn mã bên dưới -
from Bio.PopGen import GenePopPhân tích cú pháp tệp bằng phương thức GenePop.read như bên dưới:
record = GenePop.read(open("c3line.gen"))Hiển thị các locus và thông tin dân số như được cung cấp bên dưới -
>>> record.loci_list
['136255903', '136257048', '136257636']
>>> record.pop_list
['4', 'b3', '5']
>>> record.populations
[[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (3, 4), (2, 2)]),
('3', [(3, 3), (4, 4), (2, 2)]), ('4', [(3, 3), (4, 3), (None, None)])],
[('b1', [(None, None), (4, 4), (2, 2)]), ('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]), ('4',
[(None, None), (4, 4), (2, 2)]), ('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Ở đây, có sẵn ba locus trong tệp và ba tập hợp dân số: Quần thể thứ nhất có 4 bản ghi, quần thể thứ hai có 3 bản ghi và quần thể thứ ba có 5 bản ghi. record.popases hiển thị tất cả các tập hợp quần thể với dữ liệu alen cho mỗi locus.
Thao tác với tệp GenePop
Biopython cung cấp các tùy chọn để loại bỏ dữ liệu quỹ tích và dân số.
Remove a population set by position,
>>> record.remove_population(0)
>>> record.populations
[[('b1', [(None, None), (4, 4), (2, 2)]),
('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]),
('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]),
('4', [(None, None), (4, 4), (2, 2)]),
('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Remove a locus by position,
>>> record.remove_locus_by_position(0)
>>> record.loci_list
['136257048', '136257636']
>>> record.populations
[[('b1', [(4, 4), (2, 2)]), ('b2', [(4, 4), (2, 2)]), ('b3', [(4, 4), (2, 2)])],
[('1', [(4, 4), (2, 2)]), ('2', [(1, 4), (2, 2)]),
('3', [(1, 1), (2, 2)]), ('4', [(4, 4), (2, 2)]), ('5', [(4, 4), (2, 2)])]]
>>>Remove a locus by name,
>>> record.remove_locus_by_name('136257636') >>> record.loci_list
['136257048']
>>> record.populations
[[('b1', [(4, 4)]), ('b2', [(4, 4)]), ('b3', [(4, 4)])],
[('1', [(4, 4)]), ('2', [(1, 4)]),
('3', [(1, 1)]), ('4', [(4, 4)]), ('5', [(4, 4)])]]
>>>Giao diện với phần mềm GenePop
Biopython cung cấp các giao diện để tương tác với phần mềm GenePop và do đó thể hiện rất nhiều chức năng từ nó. Mô-đun Bio.PopGen.GenePop được sử dụng cho mục đích này. Một giao diện dễ sử dụng là EasyController. Hãy để chúng tôi kiểm tra cách phân tích cú pháp tệp GenePop và thực hiện một số phân tích bằng EasyController.
Đầu tiên, cài đặt phần mềm GenePop và đặt thư mục cài đặt vào đường dẫn hệ thống. Để nhận thông tin cơ bản về tệp GenePop, hãy tạo một đối tượng EasyController và sau đó gọi phương thức get_basic_info như được chỉ định bên dưới:
>>> from Bio.PopGen.GenePop.EasyController import EasyController
>>> ec = EasyController('c3line.gen')
>>> print(ec.get_basic_info())
(['4', 'b3', '5'], ['136255903', '136257048', '136257636'])
>>>Ở đây, mục đầu tiên là danh sách dân số và mục thứ hai là danh sách loci.
Để nhận danh sách tất cả các allele của một locus cụ thể, hãy gọi phương thức get_alleles_all_pops bằng cách chuyển tên locus như được chỉ định bên dưới:
>>> allele_list = ec.get_alleles_all_pops("136255903")
>>> print(allele_list)
[2, 3]Để có được danh sách alen theo quần thể và khu vực cụ thể, hãy gọi get_alleles bằng cách chuyển tên locus và vị trí của quần thể như dưới đây:
>>> allele_list = ec.get_alleles(0, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(1, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(2, "136255903")
>>> print(allele_list)
[2, 3]
>>>Tương tự, EasyController thể hiện nhiều chức năng: tần số alen, tần số kiểu gen, số liệu thống kê F đa dạng, cân bằng Hardy-Weinberg, Cân bằng bệnh liên kết, v.v.
Bộ gen là một bộ DNA hoàn chỉnh, bao gồm tất cả các gen của nó. Phân tích bộ gen đề cập đến việc nghiên cứu các gen riêng lẻ và vai trò của chúng trong việc thừa kế.
Sơ đồ bộ gen
Sơ đồ bộ gen biểu diễn thông tin di truyền dưới dạng biểu đồ. Biopython sử dụng mô-đun Bio.Graphics.GenomeDiagram để đại diện cho GenomeDiagram. Mô-đun GenomeDiagram yêu cầu phải cài đặt ReportLab.
Các bước tạo sơ đồ
Quá trình tạo một sơ đồ thường tuân theo mẫu đơn giản dưới đây:
Tạo FeatureSet cho từng bộ tính năng riêng biệt mà bạn muốn hiển thị và thêm các đối tượng Bio.SeqFeature vào chúng.
Tạo GraphSet cho từng biểu đồ bạn muốn hiển thị và thêm dữ liệu biểu đồ vào chúng.
Tạo một Bản nhạc cho mỗi bản nhạc bạn muốn trên sơ đồ và thêm GraphSets và FeatureSets vào các bản nhạc bạn yêu cầu.
Tạo một Sơ đồ và thêm các Tuyến đường vào đó.
Cho Sơ đồ vẽ hình.
Ghi hình ảnh vào một tệp.
Hãy để chúng tôi lấy một ví dụ về tệp GenBank đầu vào -
https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbkvà đọc các bản ghi từ đối tượng SeqRecord rồi cuối cùng vẽ sơ đồ bộ gen. Nó được giải thích bên dưới,
Trước tiên, chúng tôi sẽ nhập tất cả các mô-đun như hình dưới đây -
>>> from reportlab.lib import colors
>>> from reportlab.lib.units import cm
>>> from Bio.Graphics import GenomeDiagramBây giờ, nhập mô-đun SeqIO để đọc dữ liệu -
>>> from Bio import SeqIO
record = SeqIO.read("example.gb", "genbank")Ở đây, bản ghi đọc trình tự từ tệp genbank.
Bây giờ, hãy tạo một sơ đồ trống để thêm theo dõi và tập hợp tính năng -
>>> diagram = GenomeDiagram.Diagram(
"Yersinia pestis biovar Microtus plasmid pPCP1")
>>> track = diagram.new_track(1, name="Annotated Features")
>>> feature = track.new_set()Bây giờ, chúng ta có thể áp dụng các thay đổi chủ đề màu sắc bằng cách sử dụng các màu thay thế từ xanh lục sang xám như được định nghĩa bên dưới -
>>> for feature in record.features:
>>> if feature.type != "gene":
>>> continue
>>> if len(feature) % 2 == 0:
>>> color = colors.blue
>>> else:
>>> color = colors.red
>>>
>>> feature.add_feature(feature, color=color, label=True)Bây giờ bạn có thể thấy phản hồi dưới đây trên màn hình của mình -
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dc90>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dfd0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x1007627d0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57290>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57050>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57390>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57590>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57410>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57490>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d574d0>Hãy để chúng tôi vẽ sơ đồ cho các bản ghi đầu vào ở trên -
>>> diagram.draw(
format = "linear", orientation = "landscape", pagesize = 'A4',
... fragments = 4, start = 0, end = len(record))
>>> diagram.write("orchid.pdf", "PDF")
>>> diagram.write("orchid.eps", "EPS")
>>> diagram.write("orchid.svg", "SVG")
>>> diagram.write("orchid.png", "PNG")Sau khi thực hiện lệnh trên, bạn có thể thấy hình ảnh sau được lưu trong thư mục Biopython của mình.
** Result **
genome.png
Bạn cũng có thể vẽ hình ảnh ở định dạng hình tròn bằng cách thực hiện các thay đổi dưới đây:
>>> diagram.draw(
format = "circular", circular = True, pagesize = (20*cm,20*cm),
... start = 0, end = len(record), circle_core = 0.7)
>>> diagram.write("circular.pdf", "PDF")Tổng quan về nhiễm sắc thể
Phân tử DNA được đóng gói thành các cấu trúc giống như sợi chỉ được gọi là nhiễm sắc thể. Mỗi nhiễm sắc thể được tạo thành từ DNA cuộn chặt nhiều lần xung quanh các protein được gọi là histone hỗ trợ cấu trúc của nó.
Nhiễm sắc thể không thể nhìn thấy trong nhân tế bào - thậm chí không nhìn thấy dưới kính hiển vi - khi tế bào không phân chia. Tuy nhiên, DNA tạo nên nhiễm sắc thể sẽ trở nên chặt chẽ hơn trong quá trình phân chia tế bào và sau đó có thể nhìn thấy dưới kính hiển vi.
Ở người, mỗi tế bào bình thường chứa 23 cặp nhiễm sắc thể, tổng cộng là 46. Hai mươi hai trong số các cặp này, được gọi là NST thường, ở cả nam và nữ đều giống nhau. Cặp thứ 23, nhiễm sắc thể giới tính, khác nhau giữa nam và nữ. Nữ giới có hai bản sao của nhiễm sắc thể X, trong khi nam giới có một nhiễm sắc thể X và một nhiễm sắc thể Y.
Kiểu hình được định nghĩa là một đặc điểm hoặc đặc điểm quan sát được của một sinh vật chống lại một chất hóa học hoặc môi trường cụ thể. Microarray kiểu hình đồng thời đo phản ứng của một sinh vật chống lại một số lượng lớn hơn các chất hóa học & môi trường và phân tích dữ liệu để hiểu về đột biến gen, các ký tự gen, v.v.
Biopython cung cấp một mô-đun tuyệt vời, Bio.Phenotype để phân tích dữ liệu kiểu hình. Hãy để chúng tôi tìm hiểu cách phân tích cú pháp, nội suy, trích xuất và phân tích dữ liệu microarray kiểu hình trong chương này.
Phân tích cú pháp
Dữ liệu microarray kiểu hình có thể ở hai định dạng: CSV và JSON. Biopython hỗ trợ cả hai định dạng. Biopython parser phân tích cú pháp dữ liệu microarray kiểu hình và trả về dưới dạng một tập hợp các đối tượng PlateRecord. Mỗi đối tượng PlateRecord chứa một tập hợp các đối tượng WellRecord. Mỗi đối tượng WellRecord chứa dữ liệu ở định dạng 8 hàng và 12 cột. Tám hàng được biểu thị bằng A đến H và 12 cột được biểu thị bằng 01 đến 12. Ví dụ, hàng thứ 4 và cột thứ 6 được biểu thị bằng D06.
Hãy để chúng tôi hiểu định dạng và khái niệm phân tích cú pháp với ví dụ sau:
Step 1 - Tải xuống tệp Plates.csv do nhóm Biopython cung cấp - https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/Plates.csv
Step 2 - Tải mô-đun phenotpe như bên dưới -
>>> from Bio import phenotypeStep 3- Gọi phương thức phenotype.parse truyền tệp dữ liệu và tùy chọn định dạng (“pm-csv”). Nó trả về PlateRecord có thể lặp lại như bên dưới,
>>> plates = list(phenotype.parse('Plates.csv', "pm-csv"))
>>> plates
[PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'],WellRecord['A03'], ..., WellRecord['H12']')]
>>>Step 4 - Truy cập tấm đầu tiên từ danh sách như bên dưới -
>>> plate = plates[0]
>>> plate
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ...,
WellRecord['H12']')
>>>Step 5- Như đã thảo luận trước đó, một cái đĩa có 8 hàng, mỗi hàng có 12 món. WellRecord có thể được truy cập theo hai cách như được chỉ định bên dưới:
>>> well = plate["A04"]
>>> well = plate[0, 4]
>>> well WellRecord('(0.0, 0.0), (0.25, 0.0), (0.5, 0.0), (0.75, 0.0),
(1.0, 0.0), ..., (71.75, 388.0)')
>>>Step 6 - Mỗi giếng sẽ có một loạt phép đo tại các thời điểm khác nhau và có thể truy cập bằng vòng lặp for như quy định bên dưới -
>>> for v1, v2 in well:
... print(v1, v2)
...
0.0 0.0
0.25 0.0
0.5 0.0
0.75 0.0
1.0 0.0
...
71.25 388.0
71.5 388.0
71.75 388.0
>>>Nội suy
Nội suy cung cấp thông tin chi tiết hơn về dữ liệu. Biopython cung cấp các phương pháp nội suy dữ liệu WellRecord để lấy thông tin cho các mốc thời gian trung gian. Cú pháp tương tự như lập chỉ mục danh sách và do đó, rất dễ học.
Để nhận dữ liệu lúc 20,1 giờ, chỉ cần chuyển các giá trị chỉ mục như được chỉ định bên dưới:
>>> well[20.10]
69.40000000000003
>>>Chúng tôi có thể vượt qua điểm thời gian bắt đầu và điểm thời gian kết thúc cũng như được chỉ định bên dưới -
>>> well[20:30]
[67.0, 84.0, 102.0, 119.0, 135.0, 147.0, 158.0, 168.0, 179.0, 186.0]
>>>Lệnh trên nội suy dữ liệu từ 20 giờ đến 30 giờ với khoảng thời gian 1 giờ. Theo mặc định, khoảng thời gian là 1 giờ và chúng tôi có thể thay đổi nó thành bất kỳ giá trị nào. Ví dụ: hãy để chúng tôi đưa ra khoảng thời gian 15 phút (0,25 giờ) như được chỉ định bên dưới -
>>> well[20:21:0.25]
[67.0, 73.0, 75.0, 81.0]
>>>Phân tích và trích xuất
Biopython cung cấp một phương pháp phù hợp để phân tích dữ liệu WellRecord bằng cách sử dụng các hàm sigmoid của Gompertz, Logistic và Richards. Theo mặc định, phương pháp phù hợp sử dụng hàm Gompertz. Chúng ta cần gọi phương thức fit của đối tượng WellRecord để hoàn thành nhiệm vụ. Cách mã hóa như sau:
>>> well.fit()
Traceback (most recent call last):
...
Bio.MissingPythonDependencyError: Install scipy to extract curve parameters.
>>> well.model
>>> getattr(well, 'min') 0.0
>>> getattr(well, 'max') 388.0
>>> getattr(well, 'average_height')
205.42708333333334
>>>Biopython phụ thuộc vào mô-đun scipy để thực hiện phân tích nâng cao. Nó sẽ tính toán chi tiết min, max và average_height mà không cần sử dụng mô-đun scipy.
Chương này giải thích về cách vẽ các trình tự. Trước khi chuyển sang chủ đề này, chúng ta hãy hiểu những điều cơ bản về lập kế hoạch.
Âm mưu
Matplotlib là một thư viện vẽ biểu đồ Python, tạo ra các số liệu chất lượng ở nhiều định dạng. Chúng tôi có thể tạo các loại biểu đồ khác nhau như biểu đồ đường, biểu đồ, biểu đồ thanh, biểu đồ hình tròn, biểu đồ phân tán, v.v.
pyLab is a module that belongs to the matplotlib which combines the numerical module numpy with the graphical plotting module pyplot.Biopython sử dụng mô-đun pylab để vẽ các trình tự. Để thực hiện việc này, chúng ta cần nhập mã dưới đây:
import pylabTrước khi nhập, chúng ta cần cài đặt gói matplotlib bằng lệnh pip với lệnh dưới đây:
pip install matplotlibTệp đầu vào mẫu
Tạo một tệp mẫu có tên plot.fasta trong thư mục Biopython của bạn và thêm các thay đổi sau:
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF
>seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME
>seq2 EEYQTWEEFARAAEKLYLTDPMKVRVVLKYRHCDGNLCMKVTDDAVCLQYKTDQAQDVKKVEKLHGK
>seq3 MYQVWEEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVCLQYKTDQAQDV
>seq4 EEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVVSYEMRLFGVQKDNFALEHSLL
>seq5 SWEEFAKAAEVLYLEDPMKCRMCTKYRHVDHKLVVKLTDNHTVLKYVTDMAQDVKKIEKLTTLLMR
>seq6 FTNWEEFAKAAERLHSANPEKCRFVTKYNHTKGELVLKLTDDVVCLQYSTNQLQDVKKLEKLSSTLLRSI
>seq7 SWEEFVERSVQLFRGDPNATRYVMKYRHCEGKLVLKVTDDRECLKFKTDQAQDAKKMEKLNNIFF
>seq8 SWDEFVDRSVQLFRADPESTRYVMKYRHCDGKLVLKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM
>seq9 KNWEDFEIAAENMYMANPQNCRYTMKYVHSKGHILLKMSDNVKCVQYRAENMPDLKK
>seq10 FDSWDEFVSKSVELFRNHPDTTRYVVKYRHCEGKLVLKVTDNHECLKFKTDQAQDAKKMEKLô đường
Bây giờ, chúng ta hãy tạo một biểu đồ đường đơn giản cho tệp fasta ở trên.
Step 1 - Nhập mô-đun SeqIO để đọc tệp fasta.
>>> from Bio import SeqIOStep 2 - Phân tích cú pháp tệp đầu vào.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Hãy để chúng tôi nhập mô-đun pylab.
>>> import pylabStep 4 - Cấu hình biểu đồ đường bằng cách gán nhãn trục x và y.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Cấu hình biểu đồ đường bằng cách thiết lập hiển thị lưới.
>>> pylab.grid()Step 6 - Vẽ biểu đồ đường đơn giản bằng cách gọi phương thức biểu đồ và cung cấp các bản ghi làm đầu vào.
>>> pylab.plot(records)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Cuối cùng lưu biểu đồ bằng lệnh dưới đây.
>>> pylab.savefig("lines.png")Kết quả
Sau khi thực hiện lệnh trên, bạn có thể thấy hình ảnh sau được lưu trong thư mục Biopython của mình.
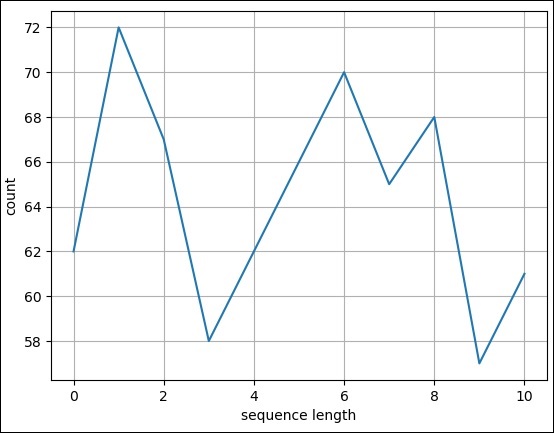
Biểu đồ Histogram
Biểu đồ được sử dụng cho dữ liệu liên tục, trong đó các thùng đại diện cho các phạm vi dữ liệu. Vẽ biểu đồ giống như biểu đồ đường ngoại trừ pylab.plot. Thay vào đó, hãy gọi phương thức hist của mô-đun pylab với các bản ghi và một số giá trị custum cho thùng (5). Mã hóa hoàn chỉnh như sau:
Step 1 - Nhập mô-đun SeqIO để đọc tệp fasta.
>>> from Bio import SeqIOStep 2 - Phân tích cú pháp tệp đầu vào.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Hãy để chúng tôi nhập mô-đun pylab.
>>> import pylabStep 4 - Cấu hình biểu đồ đường bằng cách gán nhãn trục x và y.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Cấu hình biểu đồ đường bằng cách thiết lập hiển thị lưới.
>>> pylab.grid()Step 6 - Vẽ biểu đồ đường đơn giản bằng cách gọi phương thức biểu đồ và cung cấp các bản ghi làm đầu vào.
>>> pylab.hist(records,bins=5)
(array([2., 3., 1., 3., 2.]), array([57., 60., 63., 66., 69., 72.]), <a list
of 5 Patch objects>)
>>>Step 7 - Cuối cùng lưu biểu đồ bằng lệnh dưới đây.
>>> pylab.savefig("hist.png")Kết quả
Sau khi thực hiện lệnh trên, bạn có thể thấy hình ảnh sau được lưu trong thư mục Biopython của mình.
Phần trăm GC trong trình tự
Phần trăm GC là một trong những dữ liệu phân tích thường được sử dụng để so sánh các trình tự khác nhau. Chúng ta có thể làm một biểu đồ đường đơn giản bằng cách sử dụng Phần trăm GC của một tập hợp các chuỗi và so sánh ngay lập tức. Ở đây, chúng ta chỉ có thể thay đổi dữ liệu từ độ dài chuỗi thành phần trăm GC. Mã hóa hoàn chỉnh được đưa ra dưới đây -
Step 1 - Nhập mô-đun SeqIO để đọc tệp fasta.
>>> from Bio import SeqIOStep 2 - Phân tích cú pháp tệp đầu vào.
>>> from Bio.SeqUtils import GC
>>> gc = sorted(GC(rec.seq) for rec in SeqIO.parse("plot.fasta", "fasta"))Step 3 - Hãy để chúng tôi nhập mô-đun pylab.
>>> import pylabStep 4 - Cấu hình biểu đồ đường bằng cách gán nhãn trục x và y.
>>> pylab.xlabel("Genes")
Text(0.5, 0, 'Genes')
>>> pylab.ylabel("GC Percentage")
Text(0, 0.5, 'GC Percentage')
>>>Step 5 - Cấu hình biểu đồ đường bằng cách thiết lập hiển thị lưới.
>>> pylab.grid()Step 6 - Vẽ biểu đồ đường đơn giản bằng cách gọi phương thức biểu đồ và cung cấp các bản ghi làm đầu vào.
>>> pylab.plot(gc)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Cuối cùng lưu biểu đồ bằng lệnh dưới đây.
>>> pylab.savefig("gc.png")Kết quả
Sau khi thực hiện lệnh trên, bạn có thể thấy hình ảnh sau được lưu trong thư mục Biopython của mình.
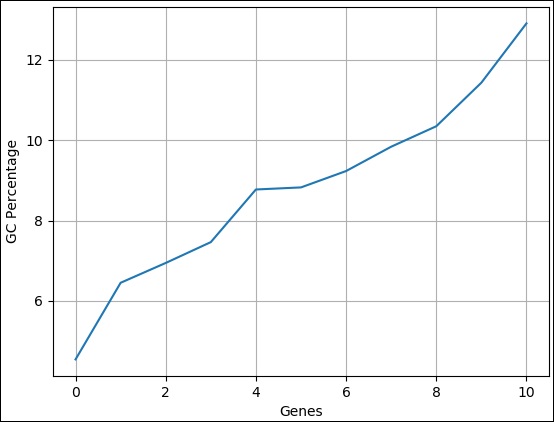
Nói chung, phân tích cụm là nhóm một tập hợp các đối tượng trong cùng một nhóm. Khái niệm này chủ yếu được sử dụng trong khai thác dữ liệu, phân tích dữ liệu thống kê, học máy, nhận dạng mẫu, phân tích hình ảnh, tin sinh học, v.v. Nó có thể đạt được bằng các thuật toán khác nhau để hiểu cách cụm được sử dụng rộng rãi trong các phân tích khác nhau.
Theo Bioinformatics, phân tích cụm chủ yếu được sử dụng trong phân tích dữ liệu biểu hiện gen để tìm các nhóm gen có biểu hiện gen tương tự.
Trong chương này, chúng ta sẽ kiểm tra các thuật toán quan trọng trong Biopython để hiểu các nguyên tắc cơ bản của việc phân cụm trên một tập dữ liệu thực.
Biopython sử dụng mô-đun Bio.Cluster để triển khai tất cả các thuật toán. Nó hỗ trợ các thuật toán sau:
- Phân cụm theo thứ bậc
- K - Phân cụm
- Bản đồ tự tổ chức
- Phân tích thành phần chính
Hãy để chúng tôi giới thiệu ngắn gọn về các thuật toán trên.
Phân cụm theo thứ bậc
Phân cụm phân cấp được sử dụng để liên kết mỗi nút bằng thước đo khoảng cách với hàng xóm gần nhất của nó và tạo một cụm. Nút Bio.Cluster có ba thuộc tính: trái, phải và khoảng cách. Hãy để chúng tôi tạo một cụm đơn giản như hình dưới đây -
>>> from Bio.Cluster import Node
>>> n = Node(1,10)
>>> n.left = 11
>>> n.right = 0
>>> n.distance = 1
>>> print(n)
(11, 0): 1Nếu bạn muốn xây dựng phân cụm dựa trên cây, hãy sử dụng lệnh dưới đây:
>>> n1 = [Node(1, 2, 0.2), Node(0, -1, 0.5)] >>> n1_tree = Tree(n1)
>>> print(n1_tree)
(1, 2): 0.2
(0, -1): 0.5
>>> print(n1_tree[0])
(1, 2): 0.2Hãy để chúng tôi thực hiện phân cụm phân cấp bằng mô-đun Bio.Cluster.
Coi khoảng cách được xác định trong một mảng.
>>> import numpy as np
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Bây giờ thêm mảng khoảng cách trong cụm cây.
>>> from Bio.Cluster import treecluster
>>> cluster = treecluster(distance)
>>> print(cluster)
(2, 1): 0.666667
(-1, 0): 9.66667Hàm trên trả về một đối tượng cụm cây. Đối tượng này chứa các nút nơi số lượng mục được nhóm lại dưới dạng hàng hoặc cột.
K - Phân cụm
Nó là một loại thuật toán phân vùng và được phân loại thành cụm k - phương tiện, phương tiện trung gian và trung gian. Hãy để chúng tôi hiểu ngắn gọn từng cụm.
K-có nghĩa là phân cụm
Cách tiếp cận này phổ biến trong khai thác dữ liệu. Mục tiêu của thuật toán này là tìm các nhóm trong dữ liệu, với số lượng nhóm được đại diện bởi biến K.
Thuật toán hoạt động lặp đi lặp lại để gán mỗi điểm dữ liệu cho một trong K nhóm dựa trên các tính năng được cung cấp. Các điểm dữ liệu được phân nhóm dựa trên sự giống nhau về tính năng.
>>> from Bio.Cluster import kcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid, error,found = kcluster(data)
>>> print(clusterid) [0 0 1]
>>> print(found)
1K-medians Clustering
Đây là một loại thuật toán phân cụm khác tính toán giá trị trung bình cho mỗi cụm để xác định trọng tâm của nó.
K-medoids Clustering
Cách tiếp cận này dựa trên một tập hợp các mục nhất định, sử dụng ma trận khoảng cách và số lượng cụm được người dùng chuyển qua.
Hãy xem xét ma trận khoảng cách như được định nghĩa dưới đây:
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Chúng ta có thể tính toán cụm k-medoids bằng lệnh dưới đây:
>>> from Bio.Cluster import kmedoids
>>> clusterid, error, found = kmedoids(distance)Chúng ta hãy xem xét một ví dụ.
Hàm kcluster lấy ma trận dữ liệu làm đầu vào chứ không phải các thể hiện Seq. Bạn cần chuyển đổi các trình tự của mình thành ma trận và cung cấp điều đó cho hàm kcluster.
Một cách để chuyển đổi dữ liệu thành ma trận chỉ chứa các phần tử số là sử dụng numpy.fromstringchức năng. Về cơ bản, nó dịch từng chữ cái theo một trình tự sang bản sao ASCII của nó.
Điều này tạo ra một mảng 2D gồm các chuỗi được mã hóa mà hàm kcluster nhận ra và sử dụng để phân cụm các chuỗi của bạn.
>>> from Bio.Cluster import kcluster
>>> import numpy as np
>>> sequence = [ 'AGCT','CGTA','AAGT','TCCG']
>>> matrix = np.asarray([np.fromstring(s, dtype=np.uint8) for s in sequence])
>>> clusterid,error,found = kcluster(matrix)
>>> print(clusterid) [1 0 0 1]Bản đồ tự tổ chức
Cách tiếp cận này là một kiểu mạng nơ-ron nhân tạo. Nó được phát triển bởi Kohonen và thường được gọi là bản đồ Kohonen. Nó tổ chức các mục thành các cụm dựa trên cấu trúc liên kết hình chữ nhật.
Hãy để chúng tôi tạo một cụm đơn giản bằng cách sử dụng cùng một khoảng cách mảng như hình dưới đây:
>>> from Bio.Cluster import somcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid,map = somcluster(data)
>>> print(map)
[[[-1.36032469 0.38667395]]
[[-0.41170578 1.35295911]]]
>>> print(clusterid)
[[1 0]
[1 0]
[1 0]]Đây, clusterid là một mảng có hai cột, trong đó số hàng bằng số lượng mục được nhóm lại và data là một mảng có kích thước là hàng hoặc cột.
Phân tích thành phần chính
Phân tích thành phần chính rất hữu ích để trực quan hóa dữ liệu chiều cao. Đây là một phương pháp sử dụng các phép toán ma trận đơn giản từ đại số tuyến tính và thống kê để tính toán một phép chiếu của dữ liệu gốc vào cùng một số hoặc ít kích thước hơn.
Phân tích thành phần chính trả về một tuple cột, tọa độ, thành phần và giá trị riêng. Chúng ta hãy xem xét những điều cơ bản của khái niệm này.
>>> from numpy import array
>>> from numpy import mean
>>> from numpy import cov
>>> from numpy.linalg import eig
# define a matrix
>>> A = array([[1, 2], [3, 4], [5, 6]])
>>> print(A)
[[1 2]
[3 4]
[5 6]]
# calculate the mean of each column
>>> M = mean(A.T, axis = 1)
>>> print(M)
[ 3. 4.]
# center columns by subtracting column means
>>> C = A - M
>>> print(C)
[[-2. -2.]
[ 0. 0.]
[ 2. 2.]]
# calculate covariance matrix of centered matrix
>>> V = cov(C.T)
>>> print(V)
[[ 4. 4.]
[ 4. 4.]]
# eigendecomposition of covariance matrix
>>> values, vectors = eig(V)
>>> print(vectors)
[[ 0.70710678 -0.70710678]
[ 0.70710678 0.70710678]]
>>> print(values)
[ 8. 0.]Hãy để chúng tôi áp dụng cùng một dữ liệu ma trận hình chữ nhật cho mô-đun Bio.Cluster như được định nghĩa bên dưới:
>>> from Bio.Cluster import pca
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> columnmean, coordinates, components, eigenvalues = pca(data)
>>> print(columnmean)
[ 3. 4.]
>>> print(coordinates)
[[-2.82842712 0. ]
[ 0. 0. ]
[ 2.82842712 0. ]]
>>> print(components)
[[ 0.70710678 0.70710678]
[ 0.70710678 -0.70710678]]
>>> print(eigenvalues)
[ 4. 0.]Tin sinh học là một lĩnh vực tuyệt vời để áp dụng các thuật toán học máy. Ở đây, chúng tôi có thông tin di truyền của một số lượng lớn sinh vật và không thể phân tích thủ công tất cả thông tin này. Nếu thuật toán học máy thích hợp được sử dụng, chúng tôi có thể trích xuất nhiều thông tin hữu ích từ những dữ liệu này. Biopython cung cấp bộ thuật toán hữu ích để thực hiện học máy có giám sát.
Việc học có giám sát dựa trên biến đầu vào (X) và biến đầu ra (Y). Nó sử dụng một thuật toán để học hàm ánh xạ từ đầu vào đến đầu ra. Nó được định nghĩa dưới đây -
Y = f(X)Mục tiêu chính của phương pháp này là ước lượng hàm ánh xạ và khi bạn có dữ liệu đầu vào mới (x), bạn có thể dự đoán các biến đầu ra (Y) cho dữ liệu đó.
Mô hình hồi quy logistic
Hồi quy logistic là một thuật toán Học máy có giám sát. Nó được sử dụng để tìm ra sự khác biệt giữa K lớp bằng cách sử dụng tổng có trọng số của các biến dự báo. Nó tính toán xác suất của một sự kiện xảy ra và có thể được sử dụng để phát hiện ung thư.
Biopython cung cấp mô-đun Bio.LogisticRegression để dự đoán các biến dựa trên thuật toán hồi quy Logistic. Hiện tại, Biopython triển khai thuật toán hồi quy logistic chỉ cho hai lớp (K = 2).
k-Những người hàng xóm gần nhất
k-Những người hàng xóm gần nhất cũng là một thuật toán học máy có giám sát. Nó hoạt động bằng cách phân loại dữ liệu dựa trên những người hàng xóm gần nhất. Biopython cung cấp mô-đun Bio.KNN để dự đoán các biến dựa trên thuật toán k-láng giềng gần nhất.
Naive Bayes
Các bộ phân loại Naive Bayes là tập hợp các thuật toán phân loại dựa trên Định lý Bayes. Nó không phải là một thuật toán đơn lẻ mà là một nhóm các thuật toán mà tất cả chúng đều có chung một nguyên tắc, tức là mọi cặp tính năng được phân loại là độc lập với nhau. Biopython cung cấp mô-đun Bio.NaiveBayes để hoạt động với thuật toán Naive Bayes.
Mô hình Markov
Mô hình Markov là một hệ thống toán học được định nghĩa là một tập hợp các biến ngẫu nhiên, trải qua quá trình chuyển đổi từ trạng thái này sang trạng thái khác theo các quy tắc xác suất nhất định. Biopython cung cấpBio.MarkovModel and Bio.HMM.MarkovModel modules to work with Markov models.
Biopython có kịch bản thử nghiệm rộng rãi để kiểm tra phần mềm trong các điều kiện khác nhau để đảm bảo rằng phần mềm không có lỗi. Để chạy tập lệnh thử nghiệm, hãy tải xuống mã nguồn của Biopython và sau đó chạy lệnh dưới đây:
python run_tests.pyThao tác này sẽ chạy tất cả các tập lệnh thử nghiệm và đưa ra kết quả sau:
Python version: 2.7.12 (v2.7.12:d33e0cf91556, Jun 26 2016, 12:10:39)
[GCC 4.2.1 (Apple Inc. build 5666) (dot 3)]
Operating system: posix darwin
test_Ace ... ok
test_Affy ... ok
test_AlignIO ... ok
test_AlignIO_ClustalIO ... ok
test_AlignIO_EmbossIO ... ok
test_AlignIO_FastaIO ... ok
test_AlignIO_MauveIO ... ok
test_AlignIO_PhylipIO ... ok
test_AlignIO_convert ... ok
...........................................
...........................................Chúng tôi cũng có thể chạy tập lệnh thử nghiệm riêng lẻ như được chỉ định bên dưới -
python test_AlignIO.pyPhần kết luận
Như chúng ta đã tìm hiểu, Biopython là một trong những phần mềm quan trọng trong lĩnh vực tin sinh học. Được viết bằng python (dễ học và viết), Nó cung cấp chức năng mở rộng để xử lý bất kỳ tính toán và hoạt động nào trong lĩnh vực tin sinh học. Nó cũng cung cấp giao diện dễ dàng và linh hoạt cho hầu hết các phần mềm tin sinh học phổ biến để khai thác các chức năng của nó.