Biopython-플로팅
이 장에서는 시퀀스를 플로팅하는 방법에 대해 설명합니다. 이 항목으로 이동하기 전에 플로팅의 기본 사항을 이해하겠습니다.
플로팅
Matplotlib는 다양한 형식으로 품질 수치를 생성하는 Python 플로팅 라이브러리입니다. 라인 차트, 히스토그램, 막대 차트, 파이 차트, 분산 형 차트 등과 같은 다양한 유형의 플롯을 만들 수 있습니다.
pyLab is a module that belongs to the matplotlib which combines the numerical module numpy with the graphical plotting module pyplot.Biopython은 시퀀스 플로팅을 위해 pylab 모듈을 사용합니다. 이렇게하려면 아래 코드를 가져와야합니다.
import pylab가져 오기 전에 아래에 주어진 명령과 함께 pip 명령을 사용하여 matplotlib 패키지를 설치해야합니다.
pip install matplotlib샘플 입력 파일
이름이 다음과 같은 샘플 파일을 만듭니다. plot.fasta Biopython 디렉토리에 다음 변경 사항을 추가하십시오.
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF
>seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME
>seq2 EEYQTWEEFARAAEKLYLTDPMKVRVVLKYRHCDGNLCMKVTDDAVCLQYKTDQAQDVKKVEKLHGK
>seq3 MYQVWEEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVCLQYKTDQAQDV
>seq4 EEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVVSYEMRLFGVQKDNFALEHSLL
>seq5 SWEEFAKAAEVLYLEDPMKCRMCTKYRHVDHKLVVKLTDNHTVLKYVTDMAQDVKKIEKLTTLLMR
>seq6 FTNWEEFAKAAERLHSANPEKCRFVTKYNHTKGELVLKLTDDVVCLQYSTNQLQDVKKLEKLSSTLLRSI
>seq7 SWEEFVERSVQLFRGDPNATRYVMKYRHCEGKLVLKVTDDRECLKFKTDQAQDAKKMEKLNNIFF
>seq8 SWDEFVDRSVQLFRADPESTRYVMKYRHCDGKLVLKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM
>seq9 KNWEDFEIAAENMYMANPQNCRYTMKYVHSKGHILLKMSDNVKCVQYRAENMPDLKK
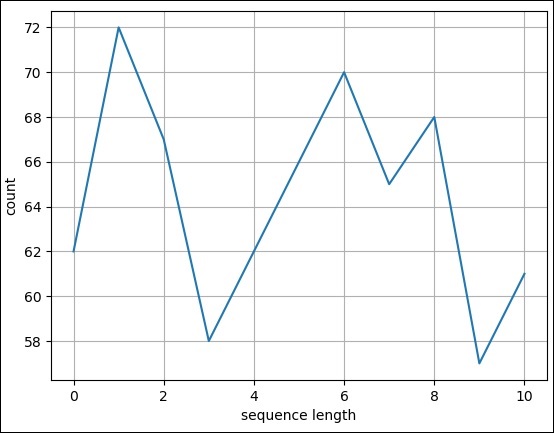
>seq10 FDSWDEFVSKSVELFRNHPDTTRYVVKYRHCEGKLVLKVTDNHECLKFKTDQAQDAKKMEK선 플롯
이제 위의 fasta 파일에 대한 간단한 라인 플롯을 생성 해 보겠습니다.
Step 1 − Fasta 파일을 읽기 위해 SeqIO 모듈을 가져옵니다.
>>> from Bio import SeqIOStep 2 − 입력 파일을 구문 분석합니다.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 − pylab 모듈을 가져 오겠습니다.
>>> import pylabStep 4 − x 및 y 축 레이블을 할당하여 라인 차트를 구성합니다.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 − 그리드 표시를 설정하여 라인 차트를 구성합니다.
>>> pylab.grid()Step 6 − plot 메서드를 호출하고 레코드를 입력으로 제공하여 간단한 선형 차트를 그립니다.
>>> pylab.plot(records)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 − 마지막으로 아래 명령을 사용하여 차트를 저장합니다.
>>> pylab.savefig("lines.png")결과
위의 명령을 실행하면 Biopython 디렉토리에 저장된 다음 이미지를 볼 수 있습니다.
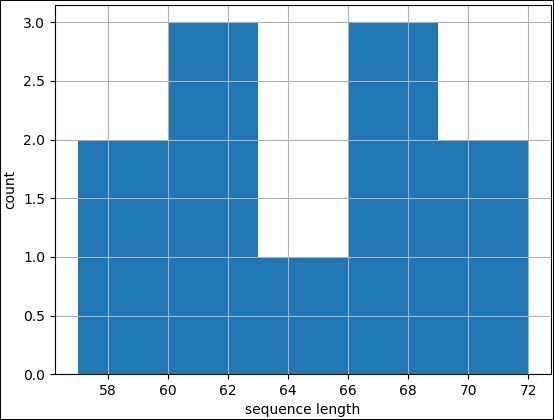
히스토그램 차트
히스토그램은 연속 데이터에 사용되며 빈은 데이터 범위를 나타냅니다. 히스토그램 그리기는 pylab.plot을 제외하고 선 차트와 동일합니다. 대신 레코드와 bin (5)에 대한 custum 값을 사용하여 pylab 모듈의 hist 메서드를 호출하십시오. 완전한 코딩은 다음과 같습니다.
Step 1 − Fasta 파일을 읽기 위해 SeqIO 모듈을 가져옵니다.
>>> from Bio import SeqIOStep 2 − 입력 파일을 구문 분석합니다.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 − pylab 모듈을 가져 오겠습니다.
>>> import pylabStep 4 − x 및 y 축 레이블을 할당하여 라인 차트를 구성합니다.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 − 그리드 표시를 설정하여 라인 차트를 구성합니다.
>>> pylab.grid()Step 6 − plot 메서드를 호출하고 레코드를 입력으로 제공하여 간단한 선형 차트를 그립니다.
>>> pylab.hist(records,bins=5)
(array([2., 3., 1., 3., 2.]), array([57., 60., 63., 66., 69., 72.]), <a list
of 5 Patch objects>)
>>>Step 7 − 마지막으로 아래 명령을 사용하여 차트를 저장합니다.
>>> pylab.savefig("hist.png")결과
위의 명령을 실행하면 Biopython 디렉토리에 저장된 다음 이미지를 볼 수 있습니다.
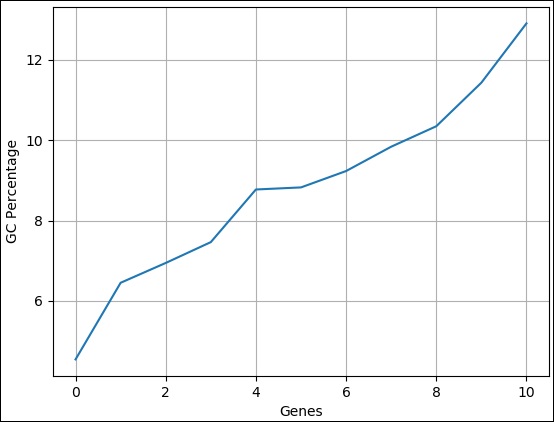
시퀀스의 GC 백분율
GC 백분율은 다른 시퀀스를 비교하기 위해 일반적으로 사용되는 분석 데이터 중 하나입니다. 시퀀스 집합의 GC 백분율을 사용하여 간단한 선형 차트를 만들고 즉시 비교할 수 있습니다. 여기서는 데이터를 시퀀스 길이에서 GC 백분율로 변경할 수 있습니다. 완전한 코딩은 다음과 같습니다.
Step 1 − Fasta 파일을 읽기 위해 SeqIO 모듈을 가져옵니다.
>>> from Bio import SeqIOStep 2 − 입력 파일을 구문 분석합니다.
>>> from Bio.SeqUtils import GC
>>> gc = sorted(GC(rec.seq) for rec in SeqIO.parse("plot.fasta", "fasta"))Step 3 − pylab 모듈을 가져 오겠습니다.
>>> import pylabStep 4 − x 및 y 축 레이블을 할당하여 라인 차트를 구성합니다.
>>> pylab.xlabel("Genes")
Text(0.5, 0, 'Genes')
>>> pylab.ylabel("GC Percentage")
Text(0, 0.5, 'GC Percentage')
>>>Step 5 − 그리드 표시를 설정하여 라인 차트를 구성합니다.
>>> pylab.grid()Step 6 − plot 메서드를 호출하고 레코드를 입력으로 제공하여 간단한 선형 차트를 그립니다.
>>> pylab.plot(gc)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 − 마지막으로 아래 명령을 사용하여 차트를 저장합니다.
>>> pylab.savefig("gc.png")결과
위의 명령을 실행하면 Biopython 디렉토리에 저장된 다음 이미지를 볼 수 있습니다.