Biopython - Guia rápido
Biopython é o maior e mais popular pacote de bioinformática para Python. Ele contém vários submódulos diferentes para tarefas comuns de bioinformática. É desenvolvido por Chapman e Chang, principalmente escrito em Python. Ele também contém código C para otimizar a parte de computação complexa do software. Ele roda em Windows, Linux, Mac OS X, etc.
Basicamente, Biopython é uma coleção de módulos python que fornecem funções para lidar com operações de DNA, RNA e sequência de proteínas, como complementação reversa de uma sequência de DNA, encontrar motivos em sequências de proteínas, etc. Ele fornece muitos analisadores para ler todos os principais bancos de dados genéticos como GenBank, SwissPort, FASTA, etc., bem como wrappers / interfaces para executar outros softwares / ferramentas de bioinformática populares como NCBI BLASTN, Entrez, etc., dentro do ambiente python. Tem projetos irmãos como BioPerl, BioJava e BioRuby.
Características
Biopython é portátil, claro e tem sintaxe fácil de aprender. Algumas das características mais importantes estão listadas abaixo -
Interpretado, interativo e orientado a objetos.
Suporta formatos relacionados a FASTA, PDB, GenBank, Blast, SCOP, PubMed / Medline, ExPASy.
Opção para lidar com formatos de sequência.
Ferramentas para gerenciar estruturas de proteínas.
BioSQL - Conjunto padrão de tabelas SQL para armazenar sequências, além de recursos e anotações.
Acesso a serviços online e banco de dados, incluindo serviços NCBI (Blast, Entrez, PubMed) e serviços ExPASY (SwissProt, Prosite).
Acesso a serviços locais, incluindo Blast, Clustalw, EMBOSS.
Metas
O objetivo do Biopython é fornecer acesso simples, padrão e abrangente à bioinformática por meio da linguagem python. Os objetivos específicos do Biopython estão listados abaixo -
Fornecimento de acesso padronizado a recursos de bioinformática.
Módulos e scripts reutilizáveis de alta qualidade.
Manipulação rápida de array que pode ser usada em código de cluster, PDB, NaiveBayes e modelo Markov.
Análise de dados genômicos.
Vantagens
Biopython requer muito menos código e apresenta as seguintes vantagens -
Fornece tipo de dados microarray usado em clustering.
Lê e grava arquivos do tipo visualização em árvore.
Suporta dados de estrutura usados para análise, representação e análise de PDB.
Suporta dados de diário usados em aplicativos Medline.
Suporta banco de dados BioSQL, que é um banco de dados padrão amplamente usado entre todos os projetos de bioinformática.
Oferece suporte ao desenvolvimento de analisador, fornecendo módulos para analisar um arquivo de bioinformática em um objeto de registro específico de formato ou uma classe genérica de sequência mais recursos.
Documentação clara baseada no estilo de livro de receitas.
Exemplo de estudo de caso
Vamos verificar alguns dos casos de uso (genética populacional, estrutura de RNA, etc.) e tentar entender como Biopython desempenha um papel importante neste campo -
Genética de População
A genética populacional é o estudo da variação genética dentro de uma população e envolve o exame e a modelagem de mudanças nas frequências de genes e alelos em populações ao longo do espaço e do tempo.
Biopython fornece módulo Bio.PopGen para genética de populações. Este módulo contém todas as funções necessárias para reunir informações sobre a genética populacional clássica.
Estrutura de RNA
As três principais macromoléculas biológicas essenciais para nossa vida são o DNA, o RNA e a proteína. As proteínas são os cavalos de batalha da célula e desempenham um papel importante como enzimas. O DNA (ácido desoxirribonucléico) é considerado o “projeto” da célula. Ele carrega todas as informações genéticas necessárias para a célula crescer, absorver nutrientes e se propagar. O RNA (ácido ribonucléico) atua como “fotocópia de DNA” na célula.
Biopython fornece objetos Bio.Sequence que representam nucleotídeos, blocos de construção de DNA e RNA.
Esta seção explica como instalar o Biopython em sua máquina. É muito fácil de instalar e não leva mais de cinco minutos.
Step 1 - Verificando a instalação do Python
Biopython foi projetado para funcionar com Python 2.5 ou versões superiores. Portanto, é obrigatório que o python seja instalado primeiro. Execute o comando abaixo em seu prompt de comando -
> python --versionÉ definido abaixo -
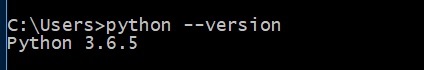
Mostra a versão do python, se instalado corretamente. Caso contrário, baixe a versão mais recente do python, instale-o e execute o comando novamente.
Step 2 - Instalação do Biopython usando pip
É fácil instalar o Biopython usando pip da linha de comando em todas as plataformas. Digite o comando abaixo -
> pip install biopythonA seguinte resposta será vista em sua tela -
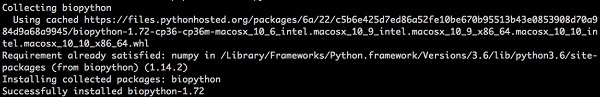
Para atualizar uma versão mais antiga do Biopython -
> pip install biopython –-upgradeA seguinte resposta será vista em sua tela -
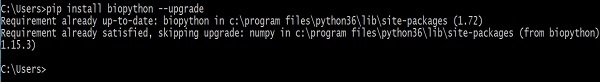
Depois de executar este comando, as versões anteriores do Biopython e do NumPy (Biopython depende dele) serão removidas antes de instalar as versões recentes.
Step 3 - Verificando a instalação do Biopython
Agora, você instalou com sucesso o Biopython em sua máquina. Para verificar se Biopython está instalado corretamente, digite o comando abaixo em seu console Python -
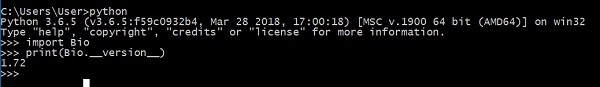
Mostra a versão do Biopython.
Alternate Way − Installing Biopython using Source
Para instalar o Biopython usando o código-fonte, siga as instruções abaixo -
Baixe o lançamento recente do Biopython no seguinte link - https://biopython.org/wiki/Download
A partir de agora, a versão mais recente é biopython-1.72.
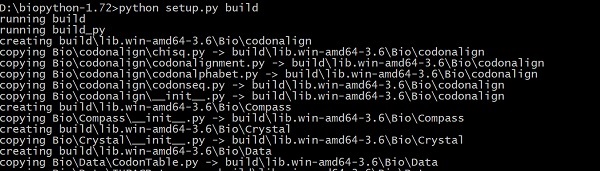
Baixe o arquivo e descompacte o arquivo compactado, mova para a pasta do código-fonte e digite o comando abaixo -
> python setup.py buildIsso irá construir Biopython a partir do código-fonte, conforme fornecido abaixo -
Agora, teste o código usando o comando abaixo -
> python setup.py test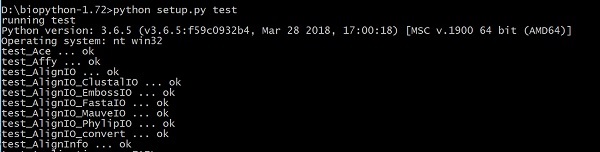
Finalmente, instale usando o comando abaixo -
> python setup.py install
Vamos criar um aplicativo Biopython simples para analisar um arquivo de bioinformática e imprimir o conteúdo. Isso nos ajudará a entender o conceito geral do Biopython e como ele ajuda no campo da bioinformática.
Step 1 - Primeiro, crie um arquivo de sequência de amostra, “example.fasta” e coloque o conteúdo abaixo nele.
>sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAV
NNFEAHTINTVVHTNDSDKGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITID
SNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTAGQYQGLVSIILTKSTTTTTTTKGT
>sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVS
NTLVGVLTLSNTSIDTVSIASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDK
NAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGNYRANITITSTIKGGGTKKGTTDKKA extensão, fasta se refere ao formato de arquivo do arquivo de sequência. O FASTA origina-se do software de bioinformática FASTA e daí seu nome. O formato FASTA tem várias sequências organizadas uma por uma e cada sequência terá seu próprio id, nome, descrição e os dados da sequência real.
Step 2 - Crie um novo script Python, * simple_example.py ", insira o código abaixo e salve-o.
from Bio.SeqIO import parse
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
file = open("example.fasta")
records = parse(file, "fasta") for record in records:
print("Id: %s" % record.id)
print("Name: %s" % record.name)
print("Description: %s" % record.description)
print("Annotations: %s" % record.annotations)
print("Sequence Data: %s" % record.seq)
print("Sequence Alphabet: %s" % record.seq.alphabet)Vamos dar uma olhada um pouco mais profunda no código -
Line 1importa a classe de análise disponível no módulo Bio.SeqIO. O módulo Bio.SeqIO é usado para ler e escrever o arquivo de sequência em diferentes formatos e a classe `parse 'é usada para analisar o conteúdo do arquivo de sequência.
Line 2importa a classe SeqRecord disponível no módulo Bio.SeqRecord. Este módulo é usado para manipular registros de sequência e a classe SeqRecord é usada para representar uma sequência particular disponível no arquivo de sequência.
*Line 3"importa a classe Seq disponível no módulo Bio.Seq. Este módulo é usado para manipular dados de sequência e a classe Seq é usada para representar os dados de sequência de um determinado registro de sequência disponível no arquivo de sequência.
Line 5 abre o arquivo “example.fasta” usando a função regular do Python, abra.
Line 7 analisa o conteúdo do arquivo de sequência e retorna o conteúdo como a lista do objeto SeqRecord.
Line 9-15 efetua loops nos registros usando python for loop e imprime os atributos do registro de sequência (SqlRecord), como id, nome, descrição, dados de sequência, etc.
Line 15 imprime o tipo da sequência usando a classe Alphabet.
Step 3 - Abra um prompt de comando e vá para a pasta que contém o arquivo de sequência, “exemplo.fasta” e execute o comando abaixo -
> python simple_example.pyStep 4- Python executa o script e imprime todos os dados de sequência disponíveis no arquivo de amostra, “example.fasta”. A saída será semelhante ao seguinte conteúdo.
Id: sp|P25730|FMS1_ECOLI
Name: sp|P25730|FMS1_ECOLI
Decription: sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
Annotations: {}
Sequence Data: MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAVNNFEAHTINTVVHTNDSD
KGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITIDSNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTA
GQYQGLVSIILTKSTTTTTTTKGT
Sequence Alphabet: SingleLetterAlphabet()
Id: sp|P15488|FMS3_ECOLI
Name: sp|P15488|FMS3_ECOLI
Decription: sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
Annotations: {}
Sequence Data: MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVSNTLVGVLTLSNTSIDTVS
IASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDKNAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGN
YRANITITSTIKGGGTKKGTTDKK
Sequence Alphabet: SingleLetterAlphabet()Vimos três classes, parse, SeqRecord e Seq neste exemplo. Essas três classes fornecem a maior parte da funcionalidade e vamos aprender essas classes na próxima seção.
Uma sequência é uma série de letras usadas para representar a proteína, DNA ou RNA de um organismo. É representado pela classe Seq. A classe Seq é definida no módulo Bio.Seq.
Vamos criar uma sequência simples no Biopython como mostrado abaixo -
>>> from Bio.Seq import Seq
>>> seq = Seq("AGCT")
>>> seq
Seq('AGCT')
>>> print(seq)
AGCTAqui, criamos uma sequência simples de proteínas AGCT e cada letra representa Alanine, Glicina, Cysteine e Threonina.
Cada objeto Seq tem dois atributos importantes -
dados - a sequência de sequência real (AGCT)
alfabeto - usado para representar o tipo de seqüência. por exemplo, sequência de DNA, sequência de RNA, etc. Por padrão, não representa nenhuma sequência e é de natureza genérica.
Módulo Alfabeto
Os objetos Seq contêm o atributo Alphabet para especificar o tipo de sequência, letras e operações possíveis. É definido no módulo Bio.Alphabet. O alfabeto pode ser definido como abaixo -
>>> from Bio.Seq import Seq
>>> myseq = Seq("AGCT")
>>> myseq
Seq('AGCT')
>>> myseq.alphabet
Alphabet()O módulo Alphabet fornece classes abaixo para representar diferentes tipos de sequências. Alfabeto - classe base para todos os tipos de alfabetos.
SingleLetterAlphabet - Alfabeto genérico com letras de tamanho um. Ele deriva de Alphabet e todos os outros tipos de alfabetos deriva dele.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import single_letter_alphabet
>>> test_seq = Seq('AGTACACTGGT', single_letter_alphabet)
>>> test_seq
Seq('AGTACACTGGT', SingleLetterAlphabet())ProteinAlphabet - Alfabeto genérico de proteína de uma única letra.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_protein
>>> test_seq = Seq('AGTACACTGGT', generic_protein)
>>> test_seq
Seq('AGTACACTGGT', ProteinAlphabet())NucleotideAlphabet - Alfabeto genérico de nucleotídeo de uma única letra.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_nucleotide
>>> test_seq = Seq('AGTACACTGGT', generic_nucleotide) >>> test_seq
Seq('AGTACACTGGT', NucleotideAlphabet())DNAAlphabet - Alfabeto genérico de DNA de uma única letra.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_dna
>>> test_seq = Seq('AGTACACTGGT', generic_dna)
>>> test_seq
Seq('AGTACACTGGT', DNAAlphabet())RNAAlphabet - Alfabeto RNA genérico de uma única letra.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_rna
>>> test_seq = Seq('AGTACACTGGT', generic_rna)
>>> test_seq
Seq('AGTACACTGGT', RNAAlphabet())O módulo Biopython, Bio.Alphabet.IUPAC fornece tipos de sequência básicos, conforme definido pela comunidade IUPAC. Ele contém as seguintes classes -
IUPACProtein (protein) - Alfabeto de proteína IUPAC de 20 aminoácidos padrão.
ExtendedIUPACProtein (extended_protein) - Alfabeto de letra única de proteína IUPAC maiúsculo estendido incluindo X.
IUPACAmbiguousDNA (ambiguous_dna) - DNA ambíguo IUPAC em maiúsculas.
IUPACUnambiguousDNA (unambiguous_dna) - DNA inequívoco IUPAC maiúsculo (GATC).
ExtendedIUPACDNA (extended_dna) - Alfabeto de DNA IUPAC estendido.
IUPACAmbiguousRNA (ambiguous_rna) - RNA ambíguo IUPAC maiúsculo.
IUPACUnambiguousRNA (unambiguous_rna) - RNA inequívoco IUPAC maiúsculo (GAUC).
Considere um exemplo simples para a classe IUPACProtein conforme mostrado abaixo -
>>> from Bio.Alphabet import IUPAC
>>> protein_seq = Seq("AGCT", IUPAC.protein)
>>> protein_seq
Seq('AGCT', IUPACProtein())
>>> protein_seq.alphabetAlém disso, Biopython expõe todos os dados de configuração relacionados à bioinformática por meio do módulo Bio.Data. Por exemplo, IUPACData.protein_letters tem as letras possíveis do alfabeto IUPACProtein.
>>> from Bio.Data import IUPACData
>>> IUPACData.protein_letters
'ACDEFGHIKLMNPQRSTVWY'Operações básicas
Esta seção explica resumidamente sobre todas as operações básicas disponíveis na classe Seq. As sequências são semelhantes às strings python. Podemos realizar operações de string Python como fatiar, contar, concatenar, localizar, dividir e separar em sequências.
Use os códigos abaixo para obter várias saídas.
To get the first value in sequence.
>>> seq_string = Seq("AGCTAGCT")
>>> seq_string[0]
'A'To print the first two values.
>>> seq_string[0:2]
Seq('AG')To print all the values.
>>> seq_string[ : ]
Seq('AGCTAGCT')To perform length and count operations.
>>> len(seq_string)
8
>>> seq_string.count('A')
2To add two sequences.
>>> from Bio.Alphabet import generic_dna, generic_protein
>>> seq1 = Seq("AGCT", generic_dna)
>>> seq2 = Seq("TCGA", generic_dna)
>>> seq1+seq2
Seq('AGCTTCGA', DNAAlphabet())Aqui, os dois objetos de sequência acima, seq1, seq2 são sequências genéricas de DNA e, portanto, você pode adicioná-los e produzir uma nova sequência. Você não pode adicionar sequências com alfabetos incompatíveis, como uma sequência de proteína e uma sequência de DNA, conforme especificado abaixo -
>>> dna_seq = Seq('AGTACACTGGT', generic_dna)
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> dna_seq + protein_seq
.....
.....
TypeError: Incompatible alphabets DNAAlphabet() and ProteinAlphabet()
>>>Para adicionar duas ou mais sequências, primeiro armazene-as em uma lista python, depois recupere-as usando 'for loop' e, finalmente, adicione-as conforme mostrado abaixo -
>>> from Bio.Alphabet import generic_dna
>>> list = [Seq("AGCT",generic_dna),Seq("TCGA",generic_dna),Seq("AAA",generic_dna)]
>>> for s in list:
... print(s)
...
AGCT
TCGA
AAA
>>> final_seq = Seq(" ",generic_dna)
>>> for s in list:
... final_seq = final_seq + s
...
>>> final_seq
Seq('AGCTTCGAAAA', DNAAlphabet())Na seção a seguir, vários códigos são fornecidos para obter resultados com base no requisito.
To change the case of sequence.
>>> from Bio.Alphabet import generic_rna
>>> rna = Seq("agct", generic_rna)
>>> rna.upper()
Seq('AGCT', RNAAlphabet())To check python membership and identity operator.
>>> rna = Seq("agct", generic_rna)
>>> 'a' in rna
True
>>> 'A' in rna
False
>>> rna1 = Seq("AGCT", generic_dna)
>>> rna is rna1
FalseTo find single letter or sequence of letter inside the given sequence.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.find('G')
1
>>> protein_seq.find('GG')
8To perform splitting operation.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.split('A')
[Seq('', ProteinAlphabet()), Seq('GU', ProteinAlphabet()),
Seq('C', ProteinAlphabet()), Seq('CUGGU', ProteinAlphabet())]To perform strip operations in the sequence.
>>> strip_seq = Seq(" AGCT ")
>>> strip_seq
Seq(' AGCT ')
>>> strip_seq.strip()
Seq('AGCT')Neste capítulo, discutiremos alguns dos recursos avançados de sequência fornecidos pelo Biopython.
Complemento e Complemento Inverso
A sequência de nucleotídeos pode ser complementada reversamente para obter uma nova sequência. Além disso, a sequência complementada pode ser complementada reversamente para obter a sequência original. Biopython fornece dois métodos para fazer essa funcionalidade -complement e reverse_complement. O código para isso é fornecido abaixo -
>>> from Bio.Alphabet import IUPAC
>>> nucleotide = Seq('TCGAAGTCAGTC', IUPAC.ambiguous_dna)
>>> nucleotide.complement()
Seq('AGCTTCAGTCAG', IUPACAmbiguousDNA())
>>>Aqui, o método complement () permite complementar uma sequência de DNA ou RNA. O método reverse_complement () complementa e reverte a sequência resultante da esquerda para a direita. É mostrado abaixo -
>>> nucleotide.reverse_complement()
Seq('GACTGACTTCGA', IUPACAmbiguousDNA())Biopython usa a variável ambiguous_dna_complement fornecida por Bio.Data.IUPACData para fazer a operação de complemento.
>>> from Bio.Data import IUPACData
>>> import pprint
>>> pprint.pprint(IUPACData.ambiguous_dna_complement) {
'A': 'T',
'B': 'V',
'C': 'G',
'D': 'H',
'G': 'C',
'H': 'D',
'K': 'M',
'M': 'K',
'N': 'N',
'R': 'Y',
'S': 'S',
'T': 'A',
'V': 'B',
'W': 'W',
'X': 'X',
'Y': 'R'}
>>>Conteúdo GC
Prevê-se que a composição da base do DNA genômico (conteúdo de GC) afete significativamente o funcionamento do genoma e a ecologia das espécies. O conteúdo do GC é o número de nucleotídeos do GC dividido pelo total de nucleotídeos.
Para obter o conteúdo do nucleotídeo GC, importe o módulo a seguir e execute as seguintes etapas -
>>> from Bio.SeqUtils import GC
>>> nucleotide = Seq("GACTGACTTCGA",IUPAC.unambiguous_dna)
>>> GC(nucleotide)
50.0Transcrição
A transcrição é o processo de transformar a sequência de DNA em sequência de RNA. O processo de transcrição biológica real está realizando um complemento reverso (TCAG → CUGA) para obter o mRNA considerando o DNA como a fita modelo. No entanto, em bioinformática e no Biopython, normalmente trabalhamos diretamente com a fita codificadora e podemos obter a sequência de mRNA alterando a letra T para U.
Um exemplo simples para o acima é o seguinte -
>>> from Bio.Seq import Seq
>>> from Bio.Seq import transcribe
>>> from Bio.Alphabet import IUPAC
>>> dna_seq = Seq("ATGCCGATCGTAT",IUPAC.unambiguous_dna) >>> transcribe(dna_seq)
Seq('AUGCCGAUCGUAU', IUPACUnambiguousRNA())
>>>Para reverter a transcrição, T é alterado para U conforme mostrado no código abaixo -
>>> rna_seq = transcribe(dna_seq)
>>> rna_seq.back_transcribe()
Seq('ATGCCGATCGTAT', IUPACUnambiguousDNA())Para obter a fita modelo de DNA, inverta_complemente o RNA transcrito de volta conforme indicado abaixo -
>>> rna_seq.back_transcribe().reverse_complement()
Seq('ATACGATCGGCAT', IUPACUnambiguousDNA())Tradução
A tradução é um processo de tradução da sequência de RNA em sequência de proteína. Considere uma sequência de RNA como mostrado abaixo -
>>> rna_seq = Seq("AUGGCCAUUGUAAU",IUPAC.unambiguous_rna)
>>> rna_seq
Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())Agora, aplique a função translate () ao código acima -
>>> rna_seq.translate()
Seq('MAIV', IUPACProtein())A seqüência de RNA acima é simples. Considere a sequência de RNA, AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA e aplique translate () -
>>> rna = Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA', IUPAC.unambiguous_rna)
>>> rna.translate()
Seq('MAIVMGR*KGAR', HasStopCodon(IUPACProtein(), '*'))Aqui, os códons de parada são indicados com um asterisco '*'.
É possível no método translate () parar no primeiro códon de parada. Para fazer isso, você pode atribuir to_stop = True em translate () da seguinte maneira -
>>> rna.translate(to_stop = True)
Seq('MAIVMGR', IUPACProtein())Aqui, o códon de parada não está incluído na sequência resultante porque não contém um.
Tabela de Tradução
A página de códigos genéticos do NCBI fornece uma lista completa das tabelas de tradução usadas pelo Biopython. Vamos ver um exemplo de tabela padrão para visualizar o código -
>>> from Bio.Data import CodonTable
>>> table = CodonTable.unambiguous_dna_by_name["Standard"]
>>> print(table)
Table 1 Standard, SGC0
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--
>>>Biopython usa esta tabela para traduzir o DNA em proteína e também para encontrar o códon de parada.
Biopython fornece um módulo, Bio.SeqIO, para ler e gravar sequências de e para um arquivo (qualquer fluxo), respectivamente. Ele suporta quase todos os formatos de arquivo disponíveis em bioinformática. A maior parte do software oferece uma abordagem diferente para diferentes formatos de arquivo. Mas, Biopython segue conscientemente uma abordagem única para apresentar os dados de sequência analisados ao usuário por meio de seu objeto SeqRecord.
Vamos aprender mais sobre SeqRecord na seção seguinte.
SeqRecord
O módulo Bio.SeqRecord fornece SeqRecord para manter as meta informações da sequência, bem como os próprios dados da sequência, conforme mostrado abaixo -
seq - É uma sequência real.
id - é o identificador principal da sequência fornecida. O tipo padrão é string.
nome - é o nome da sequência. O tipo padrão é string.
descrição - Exibe informações legíveis por humanos sobre a sequência.
anotações - É um dicionário de informações adicionais sobre a sequência.
O SeqRecord pode ser importado conforme especificado abaixo
from Bio.SeqRecord import SeqRecordVamos entender as nuances da análise do arquivo de sequência usando o arquivo de sequência real nas próximas seções.
Análise de formatos de arquivo de sequência
Esta seção explica como analisar dois dos formatos de arquivo de sequência mais populares, FASTA e GenBank.
FASTA
FASTAé o formato de arquivo mais básico para armazenar dados de sequência. Originalmente, FASTA é um pacote de software para alinhamento de sequência de DNA e proteína desenvolvido durante a evolução inicial da bioinformática e usado principalmente para pesquisar a similaridade de sequência.
Biopython fornece um arquivo FASTA de exemplo e pode ser acessado em https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Baixe e salve este arquivo em seu diretório de amostra Biopython como ‘orchid.fasta’.
O módulo Bio.SeqIO fornece o método parse () para processar arquivos de sequência e pode ser importado da seguinte forma -
from Bio.SeqIO import parseO método parse () contém dois argumentos, o primeiro é o identificador do arquivo e o segundo é o formato do arquivo.
>>> file = open('path/to/biopython/sample/orchid.fasta')
>>> for record in parse(file, "fasta"):
... print(record.id)
...
gi|2765658|emb|Z78533.1|CIZ78533
gi|2765657|emb|Z78532.1|CCZ78532
..........
..........
gi|2765565|emb|Z78440.1|PPZ78440
gi|2765564|emb|Z78439.1|PBZ78439
>>>Aqui, o método parse () retorna um objeto iterável que retorna SeqRecord em cada iteração. Sendo iterável, ele fornece muitos métodos sofisticados e fáceis e nos permite ver alguns dos recursos.
Próximo()
O método next () retorna o próximo item disponível no objeto iterável, que pode ser usado para obter a primeira sequência conforme fornecido abaixo -
>>> first_seq_record = next(SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta'))
>>> first_seq_record.id 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.name 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', SingleLetterAlphabet())
>>> first_seq_record.description 'gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> first_seq_record.annotations
{}
>>>Aqui, seq_record.annotations está vazio porque o formato FASTA não suporta anotações de sequência.
compreensão de lista
Podemos converter o objeto iterável em lista usando a compreensão de lista conforme fornecido abaixo
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> all_seq = [seq_record for seq_record in seq_iter] >>> len(all_seq)
94
>>>Aqui, usamos o método len para obter a contagem total. Podemos obter sequência com comprimento máximo da seguinte maneira -
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> max_seq = max(len(seq_record.seq) for seq_record in seq_iter)
>>> max_seq
789
>>>Podemos filtrar a sequência também usando o código abaixo -
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> seq_under_600 = [seq_record for seq_record in seq_iter if len(seq_record.seq) < 600]
>>> for seq in seq_under_600:
... print(seq.id)
...
gi|2765606|emb|Z78481.1|PIZ78481
gi|2765605|emb|Z78480.1|PGZ78480
gi|2765601|emb|Z78476.1|PGZ78476
gi|2765595|emb|Z78470.1|PPZ78470
gi|2765594|emb|Z78469.1|PHZ78469
gi|2765564|emb|Z78439.1|PBZ78439
>>>Escrever uma coleção de objetos SqlRecord (dados analisados) em um arquivo é tão simples quanto chamar o método SeqIO.write conforme abaixo -
file = open("converted.fasta", "w)
SeqIO.write(seq_record, file, "fasta")Este método pode ser usado de forma eficaz para converter o formato conforme especificado abaixo -
file = open("converted.gbk", "w)
SeqIO.write(seq_record, file, "genbank")GenBank
É um formato de sequência mais rico para genes e inclui campos para vários tipos de anotações. Biopython fornece um exemplo de arquivo GenBank e pode ser acessado emhttps://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Baixe e salve o arquivo em seu diretório de amostra Biopython como ‘orchid.gbk’
Desde então, Biopython fornece uma única função, analisar para analisar todos os formatos de bioinformática. Analisar o formato do GenBank é tão simples quanto alterar a opção de formato no método de análise.
O código para o mesmo foi fornecido abaixo -
>>> from Bio import SeqIO
>>> from Bio.SeqIO import parse
>>> seq_record = next(parse(open('path/to/biopython/sample/orchid.gbk'),'genbank'))
>>> seq_record.id
'Z78533.1'
>>> seq_record.name
'Z78533'
>>> seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', IUPACAmbiguousDNA())
>>> seq_record.description
'C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> seq_record.annotations {
'molecule_type': 'DNA',
'topology': 'linear',
'data_file_division': 'PLN',
'date': '30-NOV-2006',
'accessions': ['Z78533'],
'sequence_version': 1,
'gi': '2765658',
'keywords': ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'],
'source': 'Cypripedium irapeanum',
'organism': 'Cypripedium irapeanum',
'taxonomy': [
'Eukaryota',
'Viridiplantae',
'Streptophyta',
'Embryophyta',
'Tracheophyta',
'Spermatophyta',
'Magnoliophyta',
'Liliopsida',
'Asparagales',
'Orchidaceae',
'Cypripedioideae',
'Cypripedium'],
'references': [
Reference(title = 'Phylogenetics of the slipper orchids (Cypripedioideae:
Orchidaceae): nuclear rDNA ITS sequences', ...),
Reference(title = 'Direct Submission', ...)
]
}Sequence alignment é o processo de organizar duas ou mais sequências (de DNA, RNA ou sequências de proteínas) em uma ordem específica para identificar a região de similaridade entre elas.
Identificar a região semelhante nos permite inferir muitas informações como quais características são conservadas entre as espécies, quão próximas as espécies são geneticamente diferentes, como as espécies evoluem, etc. Biopython fornece amplo suporte para alinhamento de sequência.
Vamos aprender alguns dos recursos importantes fornecidos pelo Biopython neste capítulo -
Parsing Sequence Alignment
Biopython fornece um módulo, Bio.AlignIO, para ler e gravar alinhamentos de sequência. Em bioinformática, existem muitos formatos disponíveis para especificar os dados de alinhamento de sequência semelhantes aos dados de sequência aprendidos anteriormente. Bio.AlignIO fornece API semelhante ao Bio.SeqIO, exceto que o Bio.SeqIO funciona nos dados de sequência e Bio.AlignIO funciona nos dados de alinhamento de sequência.
Antes de começar a aprender, vamos baixar um arquivo de amostra de alinhamento de sequência da Internet.
Para baixar o arquivo de amostra, siga as etapas abaixo -
Step 1 - Abra seu navegador favorito e vá para http://pfam.xfam.org/family/browselocal na rede Internet. Ele mostrará todas as famílias Pfam em ordem alfabética.
Step 2- Escolha qualquer família com menos número de valor de semente. Ele contém dados mínimos e nos permite trabalhar facilmente com o alinhamento. Aqui, selecionamos / clicamos PF18225 e ele abre, vá parahttp://pfam.xfam.org/family/PF18225 e mostra detalhes completos sobre ele, incluindo alinhamentos de sequência.
Step 3 - Vá para a seção de alinhamento e baixe o arquivo de alinhamento de sequência no formato Estocolmo (PF18225_seed.txt).
Vamos tentar ler o arquivo de alinhamento de sequência baixado usando Bio.AlignIO como abaixo -
Módulo de importação Bio.AlignIO
>>> from Bio import AlignIOLeia o alinhamento usando o método de leitura. O método read é usado para ler os dados de alinhamento único disponíveis no arquivo fornecido. Se o arquivo fornecido contém muitos alinhamentos, podemos usar o método de análise. O método parse retorna um objeto de alinhamento iterável semelhante ao método parse no módulo Bio.SeqIO.
>>> alignment = AlignIO.read(open("PF18225_seed.txt"), "stockholm")Imprima o objeto de alinhamento.
>>> print(alignment)
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Também podemos verificar as sequências (SeqRecord) disponíveis no alinhamento, bem como abaixo -
>>> for align in alignment:
... print(align.seq)
...
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVATVANQLRGRKRRAFARHREGP
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADITA---RLDRRREHGEHGVRKKP
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMAPMLIALNYRNRESHAQVDKKP
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMAPLFKVLSFRNREDQGLVNNKP
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIMVLAPRLTAKHPYDKVQDRNRK
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVADLMRKLDLDRPFKKLERKNRT
>>>Alinhamentos Múltiplos
Em geral, a maioria dos arquivos de alinhamento de sequência contém dados de alinhamento único e é o suficiente para usar readmétodo para analisá-lo. No conceito de alinhamento de sequência múltipla, duas ou mais sequências são comparadas para melhores correspondências de subsequência entre elas e resulta em alinhamento de sequência múltipla em um único arquivo.
Se o formato de alinhamento de sequência de entrada contiver mais de um alinhamento de sequência, então precisamos usar parse método em vez de read método conforme especificado abaixo -
>>> from Bio import AlignIO
>>> alignments = AlignIO.parse(open("PF18225_seed.txt"), "stockholm")
>>> print(alignments)
<generator object parse at 0x000001CD1C7E0360>
>>> for alignment in alignments:
... print(alignment)
...
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Aqui, o método parse retorna o objeto de alinhamento iterável e pode ser iterado para obter os alinhamentos reais.
Alinhamento de sequência em pares
Pairwise sequence alignment compara apenas duas sequências por vez e fornece os melhores alinhamentos de sequência possíveis. Pairwise é fácil de entender e excepcional de inferir a partir do alinhamento de sequência resultante.
Biopython oferece um módulo especial, Bio.pairwise2para identificar a sequência de alinhamento usando o método de pares. Biopython aplica o melhor algoritmo para encontrar a sequência de alinhamento e é compatível com outro software.
Vamos escrever um exemplo para encontrar o alinhamento de sequência de duas sequências simples e hipotéticas usando o módulo de pares. Isso nos ajudará a entender o conceito de alinhamento de sequência e como programá-lo usando Biopython.
Passo 1
Importar o módulo pairwise2 com o comando dado abaixo -
>>> from Bio import pairwise2Passo 2
Crie duas sequências, seq1 e seq2 -
>>> from Bio.Seq import Seq
>>> seq1 = Seq("ACCGGT")
>>> seq2 = Seq("ACGT")etapa 3
Chame o método pairwise2.align.globalxx junto com seq1 e seq2 para encontrar os alinhamentos usando a linha de código abaixo -
>>> alignments = pairwise2.align.globalxx(seq1, seq2)Aqui, globalxxO método executa o trabalho real e encontra todos os melhores alinhamentos possíveis nas sequências fornecidas. Na verdade, Bio.pairwise2 fornece um grande conjunto de métodos que segue a convenção abaixo para encontrar alinhamentos em diferentes cenários.
<sequence alignment type>XYAqui, o tipo de alinhamento de sequência se refere ao tipo de alinhamento que pode ser global ou local. o tipo global é encontrar o alinhamento da sequência levando em consideração a sequência inteira. tipo local é encontrar o alinhamento da sequência examinando também o subconjunto das sequências fornecidas. Isso será tedioso, mas fornece uma ideia melhor sobre a semelhança entre as sequências fornecidas.
X refere-se à pontuação correspondente. Os valores possíveis são x (correspondência exata), m (pontuação baseada em caracteres idênticos), d (dicionário fornecido pelo usuário com caractere e pontuação de correspondência) e finalmente c (função definida pelo usuário para fornecer algoritmo de pontuação personalizado).
Y refere-se à penalidade de intervalo. Os valores possíveis são x (sem penalidades de gap), s (mesmas penalidades para ambas as sequências), d (penalidades diferentes para cada sequência) e finalmente c (função definida pelo usuário para fornecer penalidades de gap customizadas)
Portanto, localds também é um método válido, que encontra o alinhamento de sequência usando a técnica de alinhamento local, dicionário fornecido pelo usuário para correspondências e penalidade de intervalo fornecida pelo usuário para ambas as sequências.
>>> test_alignments = pairwise2.align.localds(seq1, seq2, blosum62, -10, -1)Aqui, blosum62 se refere a um dicionário disponível no módulo pairwise2 para fornecer a pontuação de correspondência. -10 refere-se à penalidade de abertura de gap e -1 refere-se à penalidade de extensão de gap.
Passo 4
Faça um loop sobre o objeto de alinhamento iterável, obtenha cada objeto de alinhamento individual e imprima-o.
>>> for alignment in alignments:
... print(alignment)
...
('ACCGGT', 'A-C-GT', 4.0, 0, 6)
('ACCGGT', 'AC--GT', 4.0, 0, 6)
('ACCGGT', 'A-CG-T', 4.0, 0, 6)
('ACCGGT', 'AC-G-T', 4.0, 0, 6)Etapa 5
O módulo Bio.pairwise2 fornece um método de formatação, format_alignment, para visualizar melhor o resultado -
>>> from Bio.pairwise2 import format_alignment
>>> alignments = pairwise2.align.globalxx(seq1, seq2)
>>> for alignment in alignments:
... print(format_alignment(*alignment))
...
ACCGGT
| | ||
A-C-GT
Score=4
ACCGGT
|| ||
AC--GT
Score=4
ACCGGT
| || |
A-CG-T
Score=4
ACCGGT
|| | |
AC-G-T
Score=4
>>>Biopython também fornece outro módulo para fazer o alinhamento de sequência, Alinhar. Este módulo fornece um conjunto diferente de API para simplesmente definir parâmetros como algoritmo, modo, pontuação de correspondência, penalidades de lacuna, etc. Uma olhada simples no objeto Align é a seguinte -
>>> from Bio import Align
>>> aligner = Align.PairwiseAligner()
>>> print(aligner)
Pairwise sequence aligner with parameters
match score: 1.000000
mismatch score: 0.000000
target open gap score: 0.000000
target extend gap score: 0.000000
target left open gap score: 0.000000
target left extend gap score: 0.000000
target right open gap score: 0.000000
target right extend gap score: 0.000000
query open gap score: 0.000000
query extend gap score: 0.000000
query left open gap score: 0.000000
query left extend gap score: 0.000000
query right open gap score: 0.000000
query right extend gap score: 0.000000
mode: global
>>>Suporte para ferramentas de alinhamento de sequência
Biopython fornece interface para uma série de ferramentas de alinhamento de sequência através do módulo Bio.Align.Applications. Algumas das ferramentas estão listadas abaixo -
- ClustalW
- MUSCLE
- Agulha EMBOSS e água
Vamos escrever um exemplo simples no Biopython para criar o alinhamento de sequência por meio da ferramenta de alinhamento mais popular, ClustalW.
Step 1 - Baixe o programa Clustalw de http://www.clustal.org/download/current/e instale-o. Além disso, atualize o PATH do sistema com o caminho de instalação “clustal”.
Step 2 - importe ClustalwCommanLine do módulo Bio.Align.Applications.
>>> from Bio.Align.Applications import ClustalwCommandlineStep 3 - Defina o cmd chamando ClustalwCommanLine com o arquivo de entrada, opuntia.fasta disponível no pacote Biopython. https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/opuntia.fasta
>>> cmd = ClustalwCommandline("clustalw2",
infile="/path/to/biopython/sample/opuntia.fasta")
>>> print(cmd)
clustalw2 -infile=fasta/opuntia.fastaStep 4 - Chamar cmd () executará o comando clustalw e fornecerá uma saída do arquivo de alinhamento resultante, opuntia.aln.
>>> stdout, stderr = cmd()Step 5 - Leia e imprima o arquivo de alinhamento conforme abaixo -
>>> from Bio import AlignIO
>>> align = AlignIO.read("/path/to/biopython/sample/opuntia.aln", "clustal")
>>> print(align)
SingleLetterAlphabet() alignment with 7 rows and 906 columns
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273285|gb|AF191659.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273284|gb|AF191658.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273287|gb|AF191661.1|AF191
TATACATAAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273286|gb|AF191660.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273290|gb|AF191664.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273289|gb|AF191663.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273291|gb|AF191665.1|AF191
>>>BLAST significa Basic Local Alignment Search Tool. Ele encontra regiões de similaridade entre sequências biológicas. O Biopython fornece o módulo Bio.Blast para lidar com a operação do NCBI BLAST. Você pode executar o BLAST em uma conexão local ou pela conexão com a Internet.
Vamos entender essas duas conexões resumidamente na seção a seguir -
Correndo pela Internet
Biopython fornece módulo Bio.Blast.NCBIWWW para chamar a versão online do BLAST. Para fazer isso, precisamos importar o seguinte módulo -
>>> from Bio.Blast import NCBIWWWO módulo NCBIWW fornece a função qblast para consultar a versão online do BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi. qblast suporta todos os parâmetros suportados pela versão online.
Para obter qualquer ajuda sobre este módulo, use o comando abaixo e entenda os recursos -
>>> help(NCBIWWW.qblast)
Help on function qblast in module Bio.Blast.NCBIWWW:
qblast(
program, database, sequence,
url_base = 'https://blast.ncbi.nlm.nih.gov/Blast.cgi',
auto_format = None,
composition_based_statistics = None,
db_genetic_code = None,
endpoints = None,
entrez_query = '(none)',
expect = 10.0,
filter = None,
gapcosts = None,
genetic_code = None,
hitlist_size = 50,
i_thresh = None,
layout = None,
lcase_mask = None,
matrix_name = None,
nucl_penalty = None,
nucl_reward = None,
other_advanced = None,
perc_ident = None,
phi_pattern = None,
query_file = None,
query_believe_defline = None,
query_from = None,
query_to = None,
searchsp_eff = None,
service = None,
threshold = None,
ungapped_alignment = None,
word_size = None,
alignments = 500,
alignment_view = None,
descriptions = 500,
entrez_links_new_window = None,
expect_low = None,
expect_high = None,
format_entrez_query = None,
format_object = None,
format_type = 'XML',
ncbi_gi = None,
results_file = None,
show_overview = None,
megablast = None,
template_type = None,
template_length = None
)
BLAST search using NCBI's QBLAST server or a cloud service provider.
Supports all parameters of the qblast API for Put and Get.
Please note that BLAST on the cloud supports the NCBI-BLAST Common
URL API (http://ncbi.github.io/blast-cloud/dev/api.html).
To use this feature, please set url_base to 'http://host.my.cloud.service.provider.com/cgi-bin/blast.cgi' and
format_object = 'Alignment'. For more details, please see 8. Biopython – Overview of BLAST
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE = BlastDocs&DOC_TYPE = CloudBlast
Some useful parameters:
- program blastn, blastp, blastx, tblastn, or tblastx (lower case)
- database Which database to search against (e.g. "nr").
- sequence The sequence to search.
- ncbi_gi TRUE/FALSE whether to give 'gi' identifier.
- descriptions Number of descriptions to show. Def 500.
- alignments Number of alignments to show. Def 500.
- expect An expect value cutoff. Def 10.0.
- matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45).
- filter "none" turns off filtering. Default no filtering
- format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML".
- entrez_query Entrez query to limit Blast search
- hitlist_size Number of hits to return. Default 50
- megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only)
- service plain, psi, phi, rpsblast, megablast (lower case)
This function does no checking of the validity of the parameters
and passes the values to the server as is. More help is available at:
https://ncbi.github.io/blast-cloud/dev/api.htmlNormalmente, os argumentos da função qblast são basicamente análogos a diferentes parâmetros que você pode definir na página da web do BLAST. Isso torna a função qblast fácil de entender, bem como reduz a curva de aprendizado para usá-la.
Conectando e pesquisando
Para entender o processo de conexão e busca da versão online do BLAST, vamos fazer uma busca de sequência simples (disponível em nosso arquivo de sequência local) no servidor BLAST online através do Biopython.
Step 1 - Crie um arquivo chamado blast_example.fasta no diretório Biopython e forneça as informações de sequência abaixo como entrada
Example of a single sequence in FASTA/Pearson format:
>sequence A ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattcatat
tctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtc
>sequence B ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattca
tattctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtcStep 2 - Importe o módulo NCBIWWW.
>>> from Bio.Blast import NCBIWWWStep 3 - Abra o arquivo de sequência, blast_example.fasta usando o módulo Python IO.
>>> sequence_data = open("blast_example.fasta").read()
>>> sequence_data
'Example of a single sequence in FASTA/Pearson format:\n\n\n> sequence
A\nggtaagtcctctagtacaaacacccccaatattgtgatataattaaaatt
atattcatat\ntctgttgccagaaaaaacacttttaggctatattagagccatcttctttg aagcgttgtc\n\n'Step 4- Agora, chame a função qblast passando os dados da sequência como parâmetro principal. O outro parâmetro representa o banco de dados (nt) e o programa interno (blastn).
>>> result_handle = NCBIWWW.qblast("blastn", "nt", sequence_data)
>>> result_handle
<_io.StringIO object at 0x000001EC9FAA4558>blast_resultscontém o resultado da nossa pesquisa. Ele pode ser salvo em um arquivo para uso posterior e também analisado para obter os detalhes. Aprenderemos como fazer isso na próxima seção.
Step 5 - A mesma funcionalidade pode ser feita usando o objeto Seq, ao invés de usar todo o arquivo fasta como mostrado abaixo -
>>> from Bio import SeqIO
>>> seq_record = next(SeqIO.parse(open('blast_example.fasta'),'fasta'))
>>> seq_record.id
'sequence'
>>> seq_record.seq
Seq('ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatat...gtc',
SingleLetterAlphabet())Agora, chame a função qblast passando o objeto Seq, record.seq como parâmetro principal.
>>> result_handle = NCBIWWW.qblast("blastn", "nt", seq_record.seq)
>>> print(result_handle)
<_io.StringIO object at 0x000001EC9FAA4558>O BLAST atribuirá um identificador para sua sequência automaticamente.
Step 6 - o objeto result_handle terá o resultado inteiro e pode ser salvo em um arquivo para uso posterior.
>>> with open('results.xml', 'w') as save_file:
>>> blast_results = result_handle.read()
>>> save_file.write(blast_results)Veremos como analisar o arquivo de resultado na seção posterior.
Executando BLAST autônomo
Esta seção explica como executar o BLAST no sistema local. Se você executar o BLAST no sistema local, ele pode ser mais rápido e também permite que você crie seu próprio banco de dados para pesquisar sequências.
Conectando BLAST
Em geral, executar o BLAST localmente não é recomendado devido ao seu grande tamanho, esforço extra necessário para executar o software e o custo envolvido. BLAST online é suficiente para propósitos básicos e avançados. Claro, às vezes você pode ser solicitado a instalá-lo localmente.
Considere que você está conduzindo pesquisas on-line frequentes, o que pode exigir muito tempo e um alto volume de rede e, se você tiver dados de sequência proprietários ou problemas relacionados ao IP, recomenda-se instalá-los localmente.
Para fazer isso, precisamos seguir as etapas abaixo -
Step 1- Baixe e instale o binário do blast mais recente usando o link fornecido - ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Step 2- Baixe e descompacte o banco de dados mais recente e necessário usando o link abaixo - ftp://ftp.ncbi.nlm.nih.gov/blast/db/
O software BLAST fornece muitos bancos de dados em seu site. Vamos baixar o arquivo alu.n.gz do site do banco de dados do blast e descompactá-lo na pasta alu. Este arquivo está no formato FASTA. Para usar este arquivo em nosso aplicativo blast, precisamos primeiro converter o arquivo do formato FASTA para o formato de banco de dados blast. O BLAST fornece o aplicativo makeblastdb para fazer essa conversão.
Use o snippet de código abaixo -
cd /path/to/alu
makeblastdb -in alu.n -parse_seqids -dbtype nucl -out alunA execução do código acima analisará o arquivo de entrada, alu.n, e criará o banco de dados BLAST como vários arquivos alun.nsq, alun.nsi, etc. Agora, podemos consultar esse banco de dados para encontrar a sequência.
Instalamos o BLAST em nosso servidor local e também temos um banco de dados BLAST de amostra, alun para consultar contra ele.
Step 3- Vamos criar um arquivo de sequência de amostra para consultar o banco de dados. Crie um arquivo search.fsa e coloque os dados abaixo nele.
>gnl|alu|Z15030_HSAL001056 (Alu-J)
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCT
TGAGCCTAGGAGTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAA
AGAAAAAAAAAATAGCTCTGCTGGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTG
GGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCCACGATCACACCACT
GCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
>gnl|alu|D00596_HSAL003180 (Alu-Sx)
AGCCAGGTGTGGTGGCTCACGCCTGTAATCCCACCGCTTTGGGAGGCTGAGTCAGATCAC
CTGAGGTTAGGAATTTGGGACCAGCCTGGCCAACATGGCGACACCCCAGTCTCTACTAAT
AACACAAAAAATTAGCCAGGTGTGCTGGTGCATGTCTGTAATCCCAGCTACTCAGGAGGC
TGAGGCATGAGAATTGCTCACGAGGCGGAGGTTGTAGTGAGCTGAGATCGTGGCACTGTA
CTCCAGCCTGGCGACAGAGGGAGAACCCATGTCAAAAACAAAAAAAGACACCACCAAAGG
TCAAAGCATA
>gnl|alu|X55502_HSAL000745 (Alu-J)
TGCCTTCCCCATCTGTAATTCTGGCACTTGGGGAGTCCAAGGCAGGATGATCACTTATGC
CCAAGGAATTTGAGTACCAAGCCTGGGCAATATAACAAGGCCCTGTTTCTACAAAAACTT
TAAACAATTAGCCAGGTGTGGTGGTGCGTGCCTGTGTCCAGCTACTCAGGAAGCTGAGGC
AAGAGCTTGAGGCTACAGTGAGCTGTGTTCCACCATGGTGCTCCAGCCTGGGTGACAGGG
CAAGACCCTGTCAAAAGAAAGGAAGAAAGAACGGAAGGAAAGAAGGAAAGAAACAAGGAG
AGOs dados da sequência são coletados do arquivo alu.n; portanto, corresponde ao nosso banco de dados.
Step 4 - O software BLAST fornece muitos aplicativos para pesquisar o banco de dados e usamos o blastn. blastn application requires minimum of three arguments, db, query and out. db refere-se ao banco de dados para pesquisar; query é a sequência para combinar e outé o arquivo para armazenar os resultados. Agora, execute o comando abaixo para realizar esta consulta simples -
blastn -db alun -query search.fsa -out results.xml -outfmt 5Executar o comando acima irá pesquisar e fornecer resultados no results.xml arquivo conforme fornecido abaixo (dados parcialmente) -
<?xml version = "1.0"?>
<!DOCTYPE BlastOutput PUBLIC "-//NCBI//NCBI BlastOutput/EN"
"http://www.ncbi.nlm.nih.gov/dtd/NCBI_BlastOutput.dtd">
<BlastOutput>
<BlastOutput_program>blastn</BlastOutput_program>
<BlastOutput_version>BLASTN 2.7.1+</BlastOutput_version>
<BlastOutput_reference>Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
</BlastOutput_reference>
<BlastOutput_db>alun</BlastOutput_db>
<BlastOutput_query-ID>Query_1</BlastOutput_query-ID>
<BlastOutput_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</BlastOutput_query-def>
<BlastOutput_query-len>292</BlastOutput_query-len>
<BlastOutput_param>
<Parameters>
<Parameters_expect>10</Parameters_expect>
<Parameters_sc-match>1</Parameters_sc-match>
<Parameters_sc-mismatch>-2</Parameters_sc-mismatch>
<Parameters_gap-open>0</Parameters_gap-open>
<Parameters_gap-extend>0</Parameters_gap-extend>
<Parameters_filter>L;m;</Parameters_filter>
</Parameters>
</BlastOutput_param>
<BlastOutput_iterations>
<Iteration>
<Iteration_iter-num>1</Iteration_iter-num><Iteration_query-ID>Query_1</Iteration_query-ID>
<Iteration_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</Iteration_query-def>
<Iteration_query-len>292</Iteration_query-len>
<Iteration_hits>
<Hit>
<Hit_num>1</Hit_num>
<Hit_id>gnl|alu|Z15030_HSAL001056</Hit_id>
<Hit_def>(Alu-J)</Hit_def>
<Hit_accession>Z15030_HSAL001056</Hit_accession>
<Hit_len>292</Hit_len>
<Hit_hsps>
<Hsp>
<Hsp_num>1</Hsp_num>
<Hsp_bit-score>540.342</Hsp_bit-score>
<Hsp_score>292</Hsp_score>
<Hsp_evalue>4.55414e-156</Hsp_evalue>
<Hsp_query-from>1</Hsp_query-from>
<Hsp_query-to>292</Hsp_query-to>
<Hsp_hit-from>1</Hsp_hit-from>
<Hsp_hit-to>292</Hsp_hit-to>
<Hsp_query-frame>1</Hsp_query-frame>
<Hsp_hit-frame>1</Hsp_hit-frame>
<Hsp_identity>292</Hsp_identity>
<Hsp_positive>292</Hsp_positive>
<Hsp_gaps>0</Hsp_gaps>
<Hsp_align-len>292</Hsp_align-len>
<Hsp_qseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGAGTTTG
CGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCTGGTGGTGCATG
CCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCC
ACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
</Hsp_qseq>
<Hsp_hseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGA
GTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCT
GGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGG
CTGTGGTGAGCCACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAAC
AAATAA
</Hsp_hseq>
<Hsp_midline>
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||
</Hsp_midline>
</Hsp>
</Hit_hsps>
</Hit>
.........................
.........................
.........................
</Iteration_hits>
<Iteration_stat>
<Statistics>
<Statistics_db-num>327</Statistics_db-num>
<Statistics_db-len>80506</Statistics_db-len>
<Statistics_hsp-lenv16</Statistics_hsp-len>
<Statistics_eff-space>21528364</Statistics_eff-space>
<Statistics_kappa>0.46</Statistics_kappa>
<Statistics_lambda>1.28</Statistics_lambda>
<Statistics_entropy>0.85</Statistics_entropy>
</Statistics>
</Iteration_stat>
</Iteration>
</BlastOutput_iterations>
</BlastOutput>O comando acima pode ser executado dentro do python usando o código abaixo -
>>> from Bio.Blast.Applications import NcbiblastnCommandline
>>> blastn_cline = NcbiblastnCommandline(query = "search.fasta", db = "alun",
outfmt = 5, out = "results.xml")
>>> stdout, stderr = blastn_cline()Aqui, o primeiro é um identificador para a saída de explosão e o segundo é a possível saída de erro gerada pelo comando de explosão.
Como fornecemos o arquivo de saída como argumento de linha de comando (out = “results.xml”) e definimos o formato de saída como XML (outfmt = 5), o arquivo de saída será salvo no diretório de trabalho atual.
Análise do resultado do BLAST
Geralmente, a saída do BLAST é analisada como formato XML usando o módulo NCBIXML. Para fazer isso, precisamos importar o seguinte módulo -
>>> from Bio.Blast import NCBIXMLAgora, open the file directly using python open method e use NCBIXML parse method como dado abaixo -
>>> E_VALUE_THRESH = 1e-20
>>> for record in NCBIXML.parse(open("results.xml")):
>>> if record.alignments:
>>> print("\n")
>>> print("query: %s" % record.query[:100])
>>> for align in record.alignments:
>>> for hsp in align.hsps:
>>> if hsp.expect < E_VALUE_THRESH:
>>> print("match: %s " % align.title[:100])Isso produzirá uma saída da seguinte forma -
query: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|L12964_HSAL003860 (Alu-J)
match: gnl|alu|L13042_HSAL003863 (Alu-FLA?)
match: gnl|alu|M86249_HSAL001462 (Alu-FLA?)
match: gnl|alu|M29484_HSAL002265 (Alu-J)
query: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|J03071_HSAL001860 (Alu-J)
match: gnl|alu|X72409_HSAL005025 (Alu-Sx)
query: gnl|alu|X55502_HSAL000745 (Alu-J)
match: gnl|alu|X55502_HSAL000745 (Alu-J)Entrezé um sistema de pesquisa online fornecido pelo NCBI. Ele fornece acesso a quase todos os bancos de dados de biologia molecular conhecidos com uma consulta global integrada que oferece suporte a operadores booleanos e pesquisa de campo. Ele retorna resultados de todos os bancos de dados com informações como o número de acessos de cada banco de dados, registros com links para o banco de dados de origem, etc.
Alguns dos bancos de dados populares que podem ser acessados através do Entrez estão listados abaixo -
- Pubmed
- Pubmed Central
- Nucleotídeo (banco de dados de sequência GenBank)
- Proteína (banco de dados de sequência)
- Genoma (banco de dados do genoma completo)
- Estrutura (Estrutura Macromolecular Tridimensional)
- Taxonomia (Organismos no GenBank)
- SNP (polimorfismo de nucleotídeo único)
- UniGene (clusters de sequências de transcrição orientadas por genes)
- CDD (Conserved Protein Domain Database)
- Domínios 3D (Domínios da Estrutura Entrez)
Além dos bancos de dados acima, o Entrez fornece muitos outros bancos de dados para realizar a pesquisa de campo.
Biopython fornece um módulo específico do Entrez, Bio.Entrez, para acessar o banco de dados do Entrez. Vamos aprender como acessar o Entrez usando Biopython neste capítulo -
Etapas de conexão de banco de dados
Para adicionar os recursos do Entrez, importe o seguinte módulo -
>>> from Bio import EntrezEm seguida, defina seu e-mail para identificar quem está conectado com o código fornecido abaixo -
>>> Entrez.email = '<youremail>'Em seguida, defina o parâmetro da ferramenta Entrez e, por padrão, é Biopython.
>>> Entrez.tool = 'Demoscript'Agora, call einfo function to find index term counts, last update, and available links for each database conforme definido abaixo -
>>> info = Entrez.einfo()O método einfo retorna um objeto, que fornece acesso às informações por meio de seu método de leitura, conforme mostrado abaixo -
>>> data = info.read()
>>> print(data)
<?xml version = "1.0" encoding = "UTF-8" ?>
<!DOCTYPE eInfoResult PUBLIC "-//NLM//DTD einfo 20130322//EN"
"https://eutils.ncbi.nlm.nih.gov/eutils/dtd/20130322/einfo.dtd">
<eInfoResult>
<DbList>
<DbName>pubmed</DbName>
<DbName>protein</DbName>
<DbName>nuccore</DbName>
<DbName>ipg</DbName>
<DbName>nucleotide</DbName>
<DbName>nucgss</DbName>
<DbName>nucest</DbName>
<DbName>structure</DbName>
<DbName>sparcle</DbName>
<DbName>genome</DbName>
<DbName>annotinfo</DbName>
<DbName>assembly</DbName>
<DbName>bioproject</DbName>
<DbName>biosample</DbName>
<DbName>blastdbinfo</DbName>
<DbName>books</DbName>
<DbName>cdd</DbName>
<DbName>clinvar</DbName>
<DbName>clone</DbName>
<DbName>gap</DbName>
<DbName>gapplus</DbName>
<DbName>grasp</DbName>
<DbName>dbvar</DbName>
<DbName>gene</DbName>
<DbName>gds</DbName>
<DbName>geoprofiles</DbName>
<DbName>homologene</DbName>
<DbName>medgen</DbName>
<DbName>mesh</DbName>
<DbName>ncbisearch</DbName>
<DbName>nlmcatalog</DbName>
<DbName>omim</DbName>
<DbName>orgtrack</DbName>
<DbName>pmc</DbName>
<DbName>popset</DbName>
<DbName>probe</DbName>
<DbName>proteinclusters</DbName>
<DbName>pcassay</DbName>
<DbName>biosystems</DbName>
<DbName>pccompound</DbName>
<DbName>pcsubstance</DbName>
<DbName>pubmedhealth</DbName>
<DbName>seqannot</DbName>
<DbName>snp</DbName>
<DbName>sra</DbName>
<DbName>taxonomy</DbName>
<DbName>biocollections</DbName>
<DbName>unigene</DbName>
<DbName>gencoll</DbName>
<DbName>gtr</DbName>
</DbList>
</eInfoResult>Os dados estão no formato XML e, para obter os dados como objeto python, use Entrez.read método assim que Entrez.einfo() método é invocado -
>>> info = Entrez.einfo()
>>> record = Entrez.read(info)Aqui, registro é um dicionário que tem uma chave, DbList conforme mostrado abaixo -
>>> record.keys()
[u'DbList']Acessar a chave DbList retorna a lista de nomes de banco de dados mostrada abaixo -
>>> record[u'DbList']
['pubmed', 'protein', 'nuccore', 'ipg', 'nucleotide', 'nucgss',
'nucest', 'structure', 'sparcle', 'genome', 'annotinfo', 'assembly',
'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar',
'clone', 'gap', 'gapplus', 'grasp', 'dbvar', 'gene', 'gds', 'geoprofiles',
'homologene', 'medgen', 'mesh', 'ncbisearch', 'nlmcatalog', 'omim',
'orgtrack', 'pmc', 'popset', 'probe', 'proteinclusters', 'pcassay',
'biosystems', 'pccompound', 'pcsubstance', 'pubmedhealth', 'seqannot',
'snp', 'sra', 'taxonomy', 'biocollections', 'unigene', 'gencoll', 'gtr']
>>>Basically, Entrez module parses the XML returned by Entrez search system and provide it as python dictionary and lists.
Search Database
To search any of one the Entrez databases, we can use Bio.Entrez.esearch() module. It is defined below −
>>> info = Entrez.einfo()
>>> info = Entrez.esearch(db = "pubmed",term = "genome")
>>> record = Entrez.read(info)
>>>print(record)
DictElement({u'Count': '1146113', u'RetMax': '20', u'IdList':
['30347444', '30347404', '30347317', '30347292',
'30347286', '30347249', '30347194', '30347187',
'30347172', '30347088', '30347075', '30346992',
'30346990', '30346982', '30346980', '30346969',
'30346962', '30346954', '30346941', '30346939'],
u'TranslationStack': [DictElement({u'Count':
'927819', u'Field': 'MeSH Terms', u'Term': '"genome"[MeSH Terms]',
u'Explode': 'Y'}, attributes = {})
, DictElement({u'Count': '422712', u'Field':
'All Fields', u'Term': '"genome"[All Fields]', u'Explode': 'N'}, attributes = {}),
'OR', 'GROUP'], u'TranslationSet': [DictElement({u'To': '"genome"[MeSH Terms]
OR "genome"[All Fields]', u'From': 'genome'}, attributes = {})], u'RetStart': '0',
u'QueryTranslation': '"genome"[MeSH Terms] OR "genome"[All Fields]'},
attributes = {})
>>>If you assign incorrect db then it returns
>>> info = Entrez.esearch(db = "blastdbinfo",term = "books")
>>> record = Entrez.read(info)
>>> print(record)
DictElement({u'Count': '0', u'RetMax': '0', u'IdList': [],
u'WarningList': DictElement({u'OutputMessage': ['No items found.'],
u'PhraseIgnored': [], u'QuotedPhraseNotFound': []}, attributes = {}),
u'ErrorList': DictElement({u'FieldNotFound': [], u'PhraseNotFound':
['books']}, attributes = {}), u'TranslationSet': [], u'RetStart': '0',
u'QueryTranslation': '(books[All Fields])'}, attributes = {})If you want to search across database, then you can use Entrez.egquery. This is similar to Entrez.esearch except it is enough to specify the keyword and skip the database parameter.
>>>info = Entrez.egquery(term = "entrez")
>>> record = Entrez.read(info)
>>> for row in record["eGQueryResult"]:
... print(row["DbName"], row["Count"])
...
pubmed 458
pmc 12779 mesh 1
...
...
...
biosample 7
biocollections 0Fetch Records
Enterz provides a special method, efetch to search and download the full details of a record from Entrez. Consider the following simple example −
>>> handle = Entrez.efetch(
db = "nucleotide", id = "EU490707", rettype = "fasta")Now, we can simply read the records using SeqIO object
>>> record = SeqIO.read( handle, "fasta" )
>>> record
SeqRecord(seq = Seq('ATTTTTTACGAACCTGTGGAAATTTTTGGTTATGACAATAAATCTAGTTTAGTA...GAA',
SingleLetterAlphabet()), id = 'EU490707.1', name = 'EU490707.1',
description = 'EU490707.1
Selenipedium aequinoctiale maturase K (matK) gene, partial cds; chloroplast',
dbxrefs = [])Biopython provides Bio.PDB module to manipulate polypeptide structures. The PDB (Protein Data Bank) is the largest protein structure resource available online. It hosts a lot of distinct protein structures, including protein-protein, protein-DNA, protein-RNA complexes.
In order to load the PDB, type the below command −
from Bio.PDB import *Protein Structure File Formats
The PDB distributes protein structures in three different formats −
- The XML-based file format which is not supported by Biopython
- The pdb file format, which is a specially formatted text file
- PDBx/mmCIF files format
PDB files distributed by the Protein Data Bank may contain formatting errors that make them ambiguous or difficult to parse. The Bio.PDB module attempts to deal with these errors automatically.
The Bio.PDB module implements two different parsers, one is mmCIF format and second one is pdb format.
Let us learn how to parser each of the format in detail −
mmCIF Parser
Let us download an example database in mmCIF format from pdb server using the below command −
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'mmCif')This will download the specified file (2fat.cif) from the server and store it in the current working directory.
Here, PDBList provides options to list and download files from online PDB FTP server. retrieve_pdb_file method needs the name of the file to be downloaded without extension. retrieve_pdb_file also have option to specify download directory, pdir and format of the file, file_format. The possible values of file format are as follows −
- “mmCif” (default, PDBx/mmCif file)
- “pdb” (format PDB)
- “xml” (PMDML/XML format)
- “mmtf” (highly compressed)
- “bundle” (PDB formatted archive for large structure)
To load a cif file, use Bio.MMCIF.MMCIFParser as specified below −
>>> parser = MMCIFParser(QUIET = True)
>>> data = parser.get_structure("2FAT", "2FAT.cif")Here, QUIET suppresses the warning during parsing the file. get_structure will parse the file and return the structure with id as 2FAT (first argument).
After running the above command, it parses the file and prints possible warning, if available.
Now, check the structure using the below command −
>>> data
<Structure id = 2FAT>
To get the type, use type method as specified below,
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>We have successfully parsed the file and got the structure of the protein. We will learn the details of the protein structure and how to get it in the later chapter.
PDB Parser
Let us download an example database in PDB format from pdb server using the below command −
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'pdb')This will download the specified file (pdb2fat.ent) from the server and store it in the current working directory.
To load a pdb file, use Bio.PDB.PDBParser as specified below −
>>> parser = PDBParser(PERMISSIVE = True, QUIET = True)
>>> data = parser.get_structure("2fat","pdb2fat.ent")Here, get_structure is similar to MMCIFParser. PERMISSIVE option try to parse the protein data as flexible as possible.
Now, check the structure and its type with the code snippet given below −
>>> data
<Structure id = 2fat>
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Well, the header structure stores the dictionary information. To perform this, type the below command −
>>> print(data.header.keys()) dict_keys([
'name', 'head', 'deposition_date', 'release_date', 'structure_method', 'resolution',
'structure_reference', 'journal_reference', 'author', 'compound', 'source',
'keywords', 'journal'])
>>>To get the name, use the following code −
>>> print(data.header["name"])
an anti-urokinase plasminogen activator receptor (upar) antibody: crystal
structure and binding epitope
>>>You can also check the date and resolution with the below code −
>>> print(data.header["release_date"]) 2006-11-14
>>> print(data.header["resolution"]) 1.77PDB Structure
PDB structure is composed of a single model, containing two chains.
- chain L, containing number of residues
- chain H, containing number of residues
Each residue is composed of multiple atoms, each having a 3D position represented by (x, y, z) coordinates.
Let us learn how to get the structure of the atom in detail in the below section −
Model
The Structure.get_models() method returns an iterator over the models. It is defined below −
>>> model = data.get_models()
>>> model
<generator object get_models at 0x103fa1c80>
>>> models = list(model)
>>> models [<Model id = 0>]
>>> type(models[0])
<class 'Bio.PDB.Model.Model'>Here, a Model describes exactly one 3D conformation. It contains one or more chains.
Chain
The Model.get_chain() method returns an iterator over the chains. It is defined below −
>>> chains = list(models[0].get_chains())
>>> chains
[<Chain id = L>, <Chain id = H>]
>>> type(chains[0])
<class 'Bio.PDB.Chain.Chain'>Here, Chain describes a proper polypeptide structure, i.e., a consecutive sequence of bound residues.
Residue
The Chain.get_residues() method returns an iterator over the residues. It is defined below −
>>> residue = list(chains[0].get_residues())
>>> len(residue)
293
>>> residue1 = list(chains[1].get_residues())
>>> len(residue1)
311Well, Residue holds the atoms that belong to an amino acid.
Atoms
The Residue.get_atom() returns an iterator over the atoms as defined below −
>>> atoms = list(residue[0].get_atoms())
>>> atoms
[<Atom N>, <Atom CA>, <Atom C>, <Atom Ov, <Atom CB>, <Atom CG>, <Atom OD1>, <Atom OD2>]An atom holds the 3D coordinate of an atom and it is called a Vector. It is defined below
>>> atoms[0].get_vector()
<Vector 18.49, 73.26, 44.16>It represents x, y and z co-ordinate values.
A sequence motif is a nucleotide or amino-acid sequence pattern. Sequence motifs are formed by three-dimensional arrangement of amino acids which may not be adjacent. Biopython provides a separate module, Bio.motifs to access the functionalities of sequence motif as specified below −
from Bio import motifsCreating Simple DNA Motif
Let us create a simple DNA motif sequence using the below command −
>>> from Bio import motifs
>>> from Bio.Seq import Seq
>>> DNA_motif = [ Seq("AGCT"),
... Seq("TCGA"),
... Seq("AACT"),
... ]
>>> seq = motifs.create(DNA_motif)
>>> print(seq) AGCT TCGA AACTTo count the sequence values, use the below command −
>>> print(seq.counts)
0 1 2 3
A: 2.00 1.00 0.00 1.00
C: 0.00 1.00 2.00 0.00
G: 0.00 1.00 1.00 0.00
T: 1.00 0.00 0.00 2.00Use the following code to count ‘A’ in the sequence −
>>> seq.counts["A", :]
(2, 1, 0, 1)If you want to access the columns of counts, use the below command −
>>> seq.counts[:, 3]
{'A': 1, 'C': 0, 'T': 2, 'G': 0}Creating a Sequence Logo
We shall now discuss how to create a Sequence Logo.
Consider the below sequence −
AGCTTACG
ATCGTACC
TTCCGAAT
GGTACGTA
AAGCTTGGYou can create your own logo using the following link − http://weblogo.berkeley.edu/
Add the above sequence and create a new logo and save the image named seq.png in your biopython folder.
seq.pngAfter creating the image, now run the following command −
>>> seq.weblogo("seq.png")This DNA sequence motif is represented as a sequence logo for the LexA-binding motif.
JASPAR Database
JASPAR is one of the most popular databases. It provides facilities of any of the motif formats for reading, writing and scanning sequences. It stores meta-information for each motif. The module Bio.motifs contains a specialized class jaspar.Motif to represent meta-information attributes.
It has the following notable attributes types −
- matrix_id − Unique JASPAR motif ID
- name − The name of the motif
- tf_family − The family of motif, e.g. ’Helix-Loop-Helix’
- data_type − the type of data used in motif.
Let us create a JASPAR sites format named in sample.sites in biopython folder. It is defined below −
sample.sites
>MA0001 ARNT 1
AACGTGatgtccta
>MA0001 ARNT 2
CAGGTGggatgtac
>MA0001 ARNT 3
TACGTAgctcatgc
>MA0001 ARNT 4
AACGTGacagcgct
>MA0001 ARNT 5
CACGTGcacgtcgt
>MA0001 ARNT 6
cggcctCGCGTGcIn the above file, we have created motif instances. Now, let us create a motif object from the above instances −
>>> from Bio import motifs
>>> with open("sample.sites") as handle:
... data = motifs.read(handle,"sites")
...
>>> print(data)
TF name None
Matrix ID None
Matrix:
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00Here, data reads all the motif instances from sample.sites file.
To print all the instances from data, use the below command −
>>> for instance in data.instances:
... print(instance)
...
AACGTG
CAGGTG
TACGTA
AACGTG
CACGTG
CGCGTGUse the below command to count all the values −
>>> print(data.counts)
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00
>>>BioSQL is a generic database schema designed mainly to store sequences and its related data for all RDBMS engine. It is designed in such a way that it holds the data from all popular bioinformatics databases like GenBank, Swissport, etc. It can be used to store in-house data as well.
BioSQL currently provides specific schema for the below databases −
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora/*.sql)
- SQLite (biosqldb-sqlite.sql)
It also provides minimal support for Java based HSQLDB and Derby databases.
BioPython provides very simple, easy and advanced ORM capabilities to work with BioSQL based database. BioPython provides a module, BioSQL to do the following functionality −
- Create/remove a BioSQL database
- Connect to a BioSQL database
- Parse a sequence database like GenBank, Swisport, BLAST result, Entrez result, etc., and directly load it into the BioSQL database
- Fetch the sequence data from the BioSQL database
- Fetch taxonomy data from NCBI BLAST and store it in the BioSQL database
- Run any SQL query against the BioSQL database
Overview of BioSQL Database Schema
Before going deep into the BioSQL, let us understand the basics of BioSQL schema. BioSQL schema provides 25+ tables to hold sequence data, sequence feature, sequence category/ontology and taxonomy information. Some of the important tables are as follows −
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
Creating a BioSQL Database
In this section, let us create a sample BioSQL database, biosql using the schema provided by the BioSQL team. We shall work with SQLite database as it is really easy to get started and does not have complex setup.
Here, we shall create a SQLite based BioSQL database using the below steps.
Step 1 − Download the SQLite databse engine and install it.
Step 2 − Download the BioSQL project from the GitHub URL. https://github.com/biosql/biosql
Step 3 − Open a console and create a directory using mkdir and enter into it.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 − Run the below command to create a new SQLite database.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 − Copy the biosqldb-sqlite.sql file from the BioSQL project (/sql/biosqldb-sqlite.sql`) and store it in the current directory.
Step 6 − Run the below command to create all the tables.
sqlite> .read biosqldb-sqlite.sqlNow, all tables are created in our new database.
Step 7 − Run the below command to see all the new tables in our database.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>The first three commands are configuration commands to configure SQLite to show the result in a formatted manner.
Step 8 − Copy the sample GenBank file, ls_orchid.gbk provided by BioPython team https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk into the current directory and save it as orchid.gbk.
Step 9 − Create a python script, load_orchid.py using the below code and execute it.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()The above code parses the record in the file and converts it into python objects and inserts it into BioSQL database. We will analyze the code in later section.
Finalmente, criamos um novo banco de dados BioSQL e carregamos alguns dados de amostra nele. Discutiremos as tabelas importantes no próximo capítulo.
Diagrama ER Simples
biodatabase table está no topo da hierarquia e seu objetivo principal é organizar um conjunto de dados de sequência em um único grupo / banco de dados virtual. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. Todas as tabelas relacionadas no banco de dados BioSQL têm referências à entrada de banco de dados.
bioentryA tabela contém todos os detalhes sobre uma sequência, exceto os dados da sequência. dados de sequência de um determinadobioentry será armazenado em biosequence mesa.
taxon e taxon_name são detalhes de taxonomia e cada entrada refere-se a esta tabela para especificar suas informações de taxon.
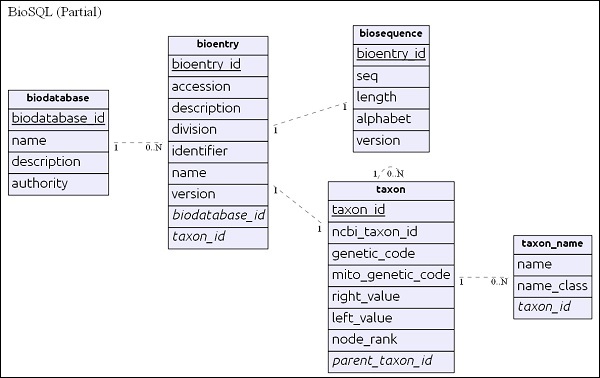
Depois de entender o esquema, vamos examinar algumas consultas na próxima seção.
Consultas BioSQL
Vamos nos aprofundar em algumas consultas SQL para entender melhor como os dados são organizados e como as tabelas estão relacionadas entre si. Antes de continuar, vamos abrir o banco de dados usando o comando abaixo e definir alguns comandos de formatação -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Você também pode usar qualquer editor SQLite para executar a consulta.
Liste o banco de dados de sequência virtual disponível no sistema conforme fornecido abaixo -
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Aqui, temos apenas um banco de dados, orchid.
Liste as entradas (top 3) disponíveis no banco de dados orchid com o código fornecido abaixo
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Liste os detalhes da sequência associados a uma entrada (acesso - Z78530, nome - C. fasciculatum 5.8S rRNA gene e ITS1 e ITS2 DNA) com o código fornecido -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Obtenha a sequência completa associada a uma entrada (acesso - Z78530, nome - C. fasciculatum 5.8S rRNA gene e ITS1 e ITS2 DNA) usando o código abaixo -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Listar táxon associado ao banco de dados bio, orquídea
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>Carregar dados no banco de dados BioSQL
Vamos aprender como carregar dados de sequência no banco de dados BioSQL neste capítulo. Já temos o código para carregar dados no banco de dados na seção anterior e o código é o seguinte -
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Teremos uma visão mais aprofundada de cada linha do código e sua finalidade -
Line 1 - Carrega o módulo SeqIO.
Line 2- Carrega o módulo BioSeqDatabase. Este módulo fornece todas as funcionalidades para interagir com o banco de dados BioSQL.
Line 3 - Carrega módulo de sistema operacional.
Line 5- open_database abre o banco de dados especificado (db) com o driver configurado (driver) e retorna um identificador para o banco de dados BioSQL (servidor). Biopython suporta bancos de dados sqlite, mysql, postgresql e oracle.
Line 6-10- o método load_database_sql carrega o sql do arquivo externo e o executa. método commit confirma a transação. Podemos pular esta etapa porque já criamos o banco de dados com o esquema.
Line 12 - métodos new_database cria um novo banco de dados virtual, orchid e retorna um manipulador db para executar o comando no banco de dados orchid.
Line 13- o método load carrega as entradas de sequência (SeqRecord iterável) no banco de dados Orchid. SqlIO.parse analisa o banco de dados GenBank e retorna todas as sequências nele como SeqRecord iterável. O segundo parâmetro (True) do método de carregamento o instrui a buscar os detalhes de taxonomia dos dados de sequência do site de explosão NCBI, se ainda não estiver disponível no sistema.
Line 14 - commit confirma a transação.
Line 15 - close fecha a conexão do banco de dados e destrói o identificador do servidor.
Obter os dados de sequência
Vamos buscar uma sequência com o identificador 2765658 no banco de dados de orquídeas como abaixo -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Aqui, o servidor ["orquídea"] retorna o identificador para buscar dados do banco de dados virtual ouchid. lookup O método fornece uma opção para selecionar sequências com base em critérios e selecionamos a sequência com o identificador 2765658. lookupretorna as informações de sequência como SeqRecordobject. Como já sabemos trabalhar com o SeqRecord`, é fácil obter dados dele.
Remover um banco de dados
Remover um banco de dados é tão simples quanto chamar o método remove_database com o nome de banco de dados adequado e, em seguida, confirmá-lo conforme especificado abaixo -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()A genética populacional desempenha um papel importante na teoria da evolução. Ele analisa a diferença genética entre as espécies, bem como dois ou mais indivíduos da mesma espécie.
Biopython fornece o módulo Bio.PopGen para genética de populações e principalmente suporta `GenePop, um pacote de genética popular desenvolvido por Michel Raymond e Francois Rousset.
Um analisador simples
Vamos escrever um aplicativo simples para analisar o formato GenePop e entender o conceito.
Baixe o arquivo genePop fornecido pela equipe Biopython no link abaixo -https://raw.githubusercontent.com/biopython/biopython/master/Tests/PopGen/c3line.gen
Carregue o módulo GenePop usando o trecho de código abaixo -
from Bio.PopGen import GenePopAnalise o arquivo usando o método GenePop.read conforme abaixo -
record = GenePop.read(open("c3line.gen"))Mostre os loci e as informações da população conforme fornecido abaixo -
>>> record.loci_list
['136255903', '136257048', '136257636']
>>> record.pop_list
['4', 'b3', '5']
>>> record.populations
[[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (3, 4), (2, 2)]),
('3', [(3, 3), (4, 4), (2, 2)]), ('4', [(3, 3), (4, 3), (None, None)])],
[('b1', [(None, None), (4, 4), (2, 2)]), ('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]), ('4',
[(None, None), (4, 4), (2, 2)]), ('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Aqui, há três loci disponíveis no arquivo e três conjuntos de população: a primeira população tem 4 registros, a segunda população tem 3 registros e a terceira população tem 5 registros. record.populations mostra todos os conjuntos de população com dados de alelos para cada locus.
Manipular o arquivo GenePop
Biopython fornece opções para remover dados de locus e população.
Remove a population set by position,
>>> record.remove_population(0)
>>> record.populations
[[('b1', [(None, None), (4, 4), (2, 2)]),
('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]),
('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]),
('4', [(None, None), (4, 4), (2, 2)]),
('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Remove a locus by position,
>>> record.remove_locus_by_position(0)
>>> record.loci_list
['136257048', '136257636']
>>> record.populations
[[('b1', [(4, 4), (2, 2)]), ('b2', [(4, 4), (2, 2)]), ('b3', [(4, 4), (2, 2)])],
[('1', [(4, 4), (2, 2)]), ('2', [(1, 4), (2, 2)]),
('3', [(1, 1), (2, 2)]), ('4', [(4, 4), (2, 2)]), ('5', [(4, 4), (2, 2)])]]
>>>Remove a locus by name,
>>> record.remove_locus_by_name('136257636') >>> record.loci_list
['136257048']
>>> record.populations
[[('b1', [(4, 4)]), ('b2', [(4, 4)]), ('b3', [(4, 4)])],
[('1', [(4, 4)]), ('2', [(1, 4)]),
('3', [(1, 1)]), ('4', [(4, 4)]), ('5', [(4, 4)])]]
>>>Interface com software GenePop
Biopython fornece interfaces para interagir com o software GenePop e, portanto, expõe muitas funcionalidades dele. O módulo Bio.PopGen.GenePop é usado para essa finalidade. Uma dessas interfaces fáceis de usar é o EasyController. Vamos verificar como analisar o arquivo GenePop e fazer algumas análises usando o EasyController.
Primeiro, instale o software GenePop e coloque a pasta de instalação no caminho do sistema. Para obter informações básicas sobre o arquivo GenePop, crie um objeto EasyController e chame o método get_basic_info conforme especificado abaixo -
>>> from Bio.PopGen.GenePop.EasyController import EasyController
>>> ec = EasyController('c3line.gen')
>>> print(ec.get_basic_info())
(['4', 'b3', '5'], ['136255903', '136257048', '136257636'])
>>>Aqui, o primeiro item é a lista de população e o segundo item é a lista de loci.
Para obter a lista de todos os alelos de um locus específico, chame o método get_alleles_all_pops passando o nome do locus conforme especificado abaixo -
>>> allele_list = ec.get_alleles_all_pops("136255903")
>>> print(allele_list)
[2, 3]Para obter a lista de alelos por população e locus específicos, chame get_alleles passando o nome do locus e a posição da população conforme fornecido abaixo -
>>> allele_list = ec.get_alleles(0, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(1, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(2, "136255903")
>>> print(allele_list)
[2, 3]
>>>Da mesma forma, EasyController expõe muitas funcionalidades: frequência de alelos, frequência de genótipos, estatísticas F multilocus, equilíbrio de Hardy-Weinberg, desequilíbrio de ligação, etc.
Um genoma é um conjunto completo de DNA, incluindo todos os seus genes. A análise do genoma se refere ao estudo de genes individuais e seus papéis na herança.
Diagrama de Genoma
O diagrama do genoma representa as informações genéticas como gráficos. Biopython usa o módulo Bio.Graphics.GenomeDiagram para representar GenomeDiagram. O módulo GenomeDiagram requer que o ReportLab seja instalado.
Etapas para criar um diagrama
O processo de criação de um diagrama geralmente segue o padrão simples abaixo -
Crie um FeatureSet para cada conjunto separado de recursos que deseja exibir e adicione objetos Bio.SeqFeature a eles.
Crie um GraphSet para cada gráfico que deseja exibir e adicione dados de gráfico a eles.
Crie uma trilha para cada trilha desejada no diagrama e adicione GraphSets e FeatureSets às trilhas de que você precisa.
Crie um diagrama e adicione as trilhas a ele.
Diga ao Diagrama para desenhar a imagem.
Grave a imagem em um arquivo.
Vamos dar um exemplo de arquivo GenBank de entrada -
https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbke ler os registros do objeto SeqRecord e, finalmente, desenhar um diagrama do genoma. É explicado abaixo,
Devemos importar todos os módulos primeiro, conforme mostrado abaixo -
>>> from reportlab.lib import colors
>>> from reportlab.lib.units import cm
>>> from Bio.Graphics import GenomeDiagramAgora, importe o módulo SeqIO para ler os dados -
>>> from Bio import SeqIO
record = SeqIO.read("example.gb", "genbank")Aqui, o registro lê a sequência do arquivo genbank.
Agora, crie um diagrama vazio para adicionar faixa e conjunto de recursos -
>>> diagram = GenomeDiagram.Diagram(
"Yersinia pestis biovar Microtus plasmid pPCP1")
>>> track = diagram.new_track(1, name="Annotated Features")
>>> feature = track.new_set()Agora, podemos aplicar alterações de tema de cores usando cores alternativas de verde a cinza, conforme definido abaixo -
>>> for feature in record.features:
>>> if feature.type != "gene":
>>> continue
>>> if len(feature) % 2 == 0:
>>> color = colors.blue
>>> else:
>>> color = colors.red
>>>
>>> feature.add_feature(feature, color=color, label=True)Agora você pode ver a resposta abaixo em sua tela -
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dc90>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dfd0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x1007627d0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57290>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57050>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57390>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57590>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57410>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57490>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d574d0>Vamos desenhar um diagrama para os registros de entrada acima -
>>> diagram.draw(
format = "linear", orientation = "landscape", pagesize = 'A4',
... fragments = 4, start = 0, end = len(record))
>>> diagram.write("orchid.pdf", "PDF")
>>> diagram.write("orchid.eps", "EPS")
>>> diagram.write("orchid.svg", "SVG")
>>> diagram.write("orchid.png", "PNG")Após executar o comando acima, você poderá ver a seguinte imagem salva em seu diretório Biopython.
** Result **
genome.png
Você também pode desenhar a imagem em formato circular, fazendo as alterações abaixo -
>>> diagram.draw(
format = "circular", circular = True, pagesize = (20*cm,20*cm),
... start = 0, end = len(record), circle_core = 0.7)
>>> diagram.write("circular.pdf", "PDF")Visão geral dos cromossomos
A molécula de DNA é empacotada em estruturas semelhantes a fios chamados cromossomos. Cada cromossomo é feito de DNA fortemente enrolado muitas vezes em torno de proteínas chamadas histonas que sustentam sua estrutura.
Os cromossomos não são visíveis no núcleo da célula - nem mesmo sob um microscópio - quando a célula não está se dividindo. No entanto, o DNA que compõe os cromossomos fica mais compactado durante a divisão celular e é então visível ao microscópio.
Em humanos, cada célula normalmente contém 23 pares de cromossomos, para um total de 46. Vinte e dois desses pares, chamados de autossomos, têm a mesma aparência em homens e mulheres. O 23º par, os cromossomos sexuais, diferem entre homens e mulheres. As mulheres têm duas cópias do cromossomo X, enquanto os homens têm um cromossomo X e um Y.
O fenótipo é definido como um caráter observável ou traço exibido por um organismo contra um determinado produto químico ou ambiente. O microarray de fenótipo mede simultaneamente a reação de um organismo contra um grande número de produtos químicos e ambientais e analisa os dados para entender a mutação do gene, caracteres do gene, etc.
Biopython fornece um excelente módulo, Bio.Phenotype, para analisar dados fenotípicos. Vamos aprender como analisar, interpolar, extrair e analisar os dados de microarray de fenótipo neste capítulo.
Análise
Os dados de microarray do fenótipo podem estar em dois formatos: CSV e JSON. Biopython suporta ambos os formatos. O analisador Biopython analisa os dados do microarray do fenótipo e retorna como uma coleção de objetos PlateRecord. Cada objeto PlateRecord contém uma coleção de objetos WellRecord. Cada objeto WellRecord contém dados em formato de 8 linhas e 12 colunas. As oito linhas são representados por A a H e 12 colunas são representadas por 01 a 12. Por exemplo, 4 th fileira e 6 th coluna são representados por D06.
Vamos entender o formato e o conceito de análise com o seguinte exemplo -
Step 1 - Baixe o arquivo Plates.csv fornecido pela equipe Biopython - https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/Plates.csv
Step 2 - Carregue o módulo de fenótipo conforme abaixo -
>>> from Bio import phenotypeStep 3- Invoque o método phenotype.parse passando o arquivo de dados e a opção de formato (“pm-csv”). Ele retorna o PlateRecord iterável como abaixo,
>>> plates = list(phenotype.parse('Plates.csv', "pm-csv"))
>>> plates
[PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'],WellRecord['A03'], ..., WellRecord['H12']')]
>>>Step 4 - Acesse a primeira placa da lista conforme abaixo -
>>> plate = plates[0]
>>> plate
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ...,
WellRecord['H12']')
>>>Step 5- Conforme discutido anteriormente, um prato contém 8 linhas, cada uma com 12 itens. WellRecord pode ser acessado de duas maneiras, conforme especificado abaixo -
>>> well = plate["A04"]
>>> well = plate[0, 4]
>>> well WellRecord('(0.0, 0.0), (0.25, 0.0), (0.5, 0.0), (0.75, 0.0),
(1.0, 0.0), ..., (71.75, 388.0)')
>>>Step 6 - Cada poço terá uma série de medições em pontos de tempo diferentes e pode ser acessado usando o loop for conforme especificado abaixo -
>>> for v1, v2 in well:
... print(v1, v2)
...
0.0 0.0
0.25 0.0
0.5 0.0
0.75 0.0
1.0 0.0
...
71.25 388.0
71.5 388.0
71.75 388.0
>>>Interpolação
A interpolação fornece mais informações sobre os dados. Biopython fornece métodos para interpolar dados WellRecord para obter informações para pontos de tempo intermediários. A sintaxe é semelhante à indexação de lista e, portanto, fácil de aprender.
Para obter os dados em 20,1 horas, basta passar como valores de índice conforme especificado abaixo -
>>> well[20.10]
69.40000000000003
>>>Podemos passar o ponto de tempo de início e ponto de tempo de término, bem como especificado abaixo -
>>> well[20:30]
[67.0, 84.0, 102.0, 119.0, 135.0, 147.0, 158.0, 168.0, 179.0, 186.0]
>>>O comando acima interpola dados de 20 a 30 horas com intervalo de 1 hora. Por padrão, o intervalo é de 1 hora e podemos alterá-lo para qualquer valor. Por exemplo, vamos dar um intervalo de 15 minutos (0,25 hora) conforme especificado abaixo -
>>> well[20:21:0.25]
[67.0, 73.0, 75.0, 81.0]
>>>Analisar e Extrair
Biopython fornece um método adequado para analisar os dados do WellRecord usando as funções sigmóide de Gompertz, Logística e Richards. Por padrão, o método de ajuste usa a função Gompertz. Precisamos chamar o método fit do objeto WellRecord para realizar a tarefa. A codificação é a seguinte -
>>> well.fit()
Traceback (most recent call last):
...
Bio.MissingPythonDependencyError: Install scipy to extract curve parameters.
>>> well.model
>>> getattr(well, 'min') 0.0
>>> getattr(well, 'max') 388.0
>>> getattr(well, 'average_height')
205.42708333333334
>>>Biopython depende do módulo scipy para fazer análises avançadas. Ele irá calcular os detalhes min, max e average_height sem usar o módulo scipy.
Este capítulo explica como plotar sequências. Antes de passar para este tópico, vamos entender os fundamentos da plotagem.
Plotagem
Matplotlib é uma biblioteca de plotagem Python que produz figuras de qualidade em uma variedade de formatos. Podemos criar diferentes tipos de gráficos, como gráfico de linha, histogramas, gráfico de barras, gráfico de pizza, gráfico de dispersão, etc.
pyLab is a module that belongs to the matplotlib which combines the numerical module numpy with the graphical plotting module pyplot.Biopython usa módulo pylab para plotar sequências. Para fazer isso, precisamos importar o código abaixo -
import pylabAntes de importar, precisamos instalar o pacote matplotlib usando o comando pip com o comando fornecido abaixo -
pip install matplotlibArquivo de entrada de amostra
Crie um arquivo de amostra chamado plot.fasta em seu diretório Biopython e adicione as seguintes alterações -
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF
>seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME
>seq2 EEYQTWEEFARAAEKLYLTDPMKVRVVLKYRHCDGNLCMKVTDDAVCLQYKTDQAQDVKKVEKLHGK
>seq3 MYQVWEEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVCLQYKTDQAQDV
>seq4 EEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVVSYEMRLFGVQKDNFALEHSLL
>seq5 SWEEFAKAAEVLYLEDPMKCRMCTKYRHVDHKLVVKLTDNHTVLKYVTDMAQDVKKIEKLTTLLMR
>seq6 FTNWEEFAKAAERLHSANPEKCRFVTKYNHTKGELVLKLTDDVVCLQYSTNQLQDVKKLEKLSSTLLRSI
>seq7 SWEEFVERSVQLFRGDPNATRYVMKYRHCEGKLVLKVTDDRECLKFKTDQAQDAKKMEKLNNIFF
>seq8 SWDEFVDRSVQLFRADPESTRYVMKYRHCDGKLVLKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM
>seq9 KNWEDFEIAAENMYMANPQNCRYTMKYVHSKGHILLKMSDNVKCVQYRAENMPDLKK
>seq10 FDSWDEFVSKSVELFRNHPDTTRYVVKYRHCEGKLVLKVTDNHECLKFKTDQAQDAKKMEKGráfico de linha
Agora, vamos criar um gráfico de linha simples para o arquivo fasta acima.
Step 1 - Importe o módulo SeqIO para ler o arquivo fasta.
>>> from Bio import SeqIOStep 2 - Analise o arquivo de entrada.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Vamos importar o módulo pylab.
>>> import pylabStep 4 - Configure o gráfico de linha atribuindo rótulos dos eixos xey.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Configure o gráfico de linha definindo a exibição da grade.
>>> pylab.grid()Step 6 - Desenhe um gráfico de linha simples chamando o método de plotagem e fornecendo registros como entrada.
>>> pylab.plot(records)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Finalmente salve o gráfico usando o comando abaixo.
>>> pylab.savefig("lines.png")Resultado
Após executar o comando acima, você poderá ver a seguinte imagem salva em seu diretório Biopython.
Gráfico de Histograma
Um histograma é usado para dados contínuos, em que os compartimentos representam intervalos de dados. O desenho do histograma é igual ao gráfico de linha, exceto pylab.plot. Em vez disso, chame o método hist do módulo pylab com registros e algum valor de custum para as caixas (5). A codificação completa é a seguinte -
Step 1 - Importe o módulo SeqIO para ler o arquivo fasta.
>>> from Bio import SeqIOStep 2 - Analise o arquivo de entrada.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Vamos importar o módulo pylab.
>>> import pylabStep 4 - Configure o gráfico de linha atribuindo rótulos dos eixos xey.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Configure o gráfico de linha definindo a exibição da grade.
>>> pylab.grid()Step 6 - Desenhe um gráfico de linha simples chamando o método de plotagem e fornecendo registros como entrada.
>>> pylab.hist(records,bins=5)
(array([2., 3., 1., 3., 2.]), array([57., 60., 63., 66., 69., 72.]), <a list
of 5 Patch objects>)
>>>Step 7 - Finalmente salve o gráfico usando o comando abaixo.
>>> pylab.savefig("hist.png")Resultado
Após executar o comando acima, você poderá ver a seguinte imagem salva em seu diretório Biopython.
Porcentagem GC na sequência
A porcentagem de GC é um dos dados analíticos comumente usados para comparar diferentes sequências. Podemos fazer um gráfico de linha simples usando a porcentagem GC de um conjunto de sequências e compará-lo imediatamente. Aqui, podemos apenas alterar os dados do comprimento da sequência para a porcentagem de GC. A codificação completa é fornecida abaixo -
Step 1 - Importe o módulo SeqIO para ler o arquivo fasta.
>>> from Bio import SeqIOStep 2 - Analise o arquivo de entrada.
>>> from Bio.SeqUtils import GC
>>> gc = sorted(GC(rec.seq) for rec in SeqIO.parse("plot.fasta", "fasta"))Step 3 - Vamos importar o módulo pylab.
>>> import pylabStep 4 - Configure o gráfico de linha atribuindo rótulos dos eixos xey.
>>> pylab.xlabel("Genes")
Text(0.5, 0, 'Genes')
>>> pylab.ylabel("GC Percentage")
Text(0, 0.5, 'GC Percentage')
>>>Step 5 - Configure o gráfico de linha definindo a exibição da grade.
>>> pylab.grid()Step 6 - Desenhe um gráfico de linha simples chamando o método de plotagem e fornecendo registros como entrada.
>>> pylab.plot(gc)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Finalmente salve o gráfico usando o comando abaixo.
>>> pylab.savefig("gc.png")Resultado
Após executar o comando acima, você poderá ver a seguinte imagem salva em seu diretório Biopython.
Em geral, a análise de cluster agrupa um conjunto de objetos no mesmo grupo. Este conceito é usado principalmente em mineração de dados, análise estatística de dados, aprendizado de máquina, reconhecimento de padrões, análise de imagens, bioinformática, etc. Ele pode ser alcançado por vários algoritmos para entender como o cluster é amplamente utilizado em diferentes análises.
De acordo com a Bioinformática, a análise de agrupamento é usada principalmente na análise de dados de expressão gênica para encontrar grupos de genes com expressão gênica semelhante.
Neste capítulo, verificaremos algoritmos importantes no Biopython para entender os fundamentos do agrupamento em um conjunto de dados real.
Biopython usa o módulo Bio.Cluster para implementar todos os algoritmos. Ele suporta os seguintes algoritmos -
- Agrupamento hierárquico
- K - Clustering
- Mapas auto-organizáveis
- Análise do componente principal
Vamos fazer uma breve introdução sobre os algoritmos acima.
Agrupamento hierárquico
O clustering hierárquico é usado para ligar cada nó por uma medida de distância ao seu vizinho mais próximo e criar um cluster. O nó Bio.Cluster possui três atributos: esquerda, direita e distância. Vamos criar um cluster simples como mostrado abaixo -
>>> from Bio.Cluster import Node
>>> n = Node(1,10)
>>> n.left = 11
>>> n.right = 0
>>> n.distance = 1
>>> print(n)
(11, 0): 1Se você quiser construir um clustering baseado em árvore, use o comando abaixo -
>>> n1 = [Node(1, 2, 0.2), Node(0, -1, 0.5)] >>> n1_tree = Tree(n1)
>>> print(n1_tree)
(1, 2): 0.2
(0, -1): 0.5
>>> print(n1_tree[0])
(1, 2): 0.2Vamos fazer clustering hierárquico usando o módulo Bio.Cluster.
Considere que a distância é definida em uma matriz.
>>> import numpy as np
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Agora adicione a matriz de distância no cluster de árvore.
>>> from Bio.Cluster import treecluster
>>> cluster = treecluster(distance)
>>> print(cluster)
(2, 1): 0.666667
(-1, 0): 9.66667A função acima retorna um objeto Tree cluster. Este objeto contém nós onde o número de itens são agrupados como linhas ou colunas.
K - Clustering
É um tipo de algoritmo de particionamento e classificado em k - médias, medianas e agrupamento de medoides. Vamos entender cada um dos agrupamentos resumidamente.
Clustering K-means
Essa abordagem é popular na mineração de dados. O objetivo deste algoritmo é encontrar grupos nos dados, com o número de grupos representado pela variável K.
O algoritmo funciona iterativamente para atribuir cada ponto de dados a um dos grupos K com base nos recursos que são fornecidos. Os pontos de dados são agrupados com base na similaridade de recursos.
>>> from Bio.Cluster import kcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid, error,found = kcluster(data)
>>> print(clusterid) [0 0 1]
>>> print(found)
1Agrupamento de K-medianas
É outro tipo de algoritmo de agrupamento que calcula a média de cada agrupamento para determinar seu centróide.
Agrupamento de K-medoides
Essa abordagem é baseada em um determinado conjunto de itens, utilizando a matriz de distância e o número de clusters passados pelo usuário.
Considere a matriz de distância conforme definido abaixo -
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Podemos calcular o agrupamento de k-medoides usando o comando abaixo -
>>> from Bio.Cluster import kmedoids
>>> clusterid, error, found = kmedoids(distance)Vamos considerar um exemplo.
A função kcluster recebe uma matriz de dados como entrada e não instâncias Seq. Você precisa converter suas sequências em uma matriz e fornecer isso para a função kcluster.
Uma maneira de converter os dados em uma matriz contendo apenas elementos numéricos é usando o numpy.fromstringfunção. Basicamente, ele traduz cada letra em uma sequência para sua contraparte ASCII.
Isso cria uma matriz 2D de sequências codificadas que a função kcluster reconheceu e usa para agrupar suas sequências.
>>> from Bio.Cluster import kcluster
>>> import numpy as np
>>> sequence = [ 'AGCT','CGTA','AAGT','TCCG']
>>> matrix = np.asarray([np.fromstring(s, dtype=np.uint8) for s in sequence])
>>> clusterid,error,found = kcluster(matrix)
>>> print(clusterid) [1 0 0 1]Mapas auto-organizáveis
Essa abordagem é um tipo de rede neural artificial. Ele é desenvolvido por Kohonen e frequentemente chamado de mapa de Kohonen. Ele organiza itens em clusters com base na topologia retangular.
Vamos criar um cluster simples usando a mesma distância de matriz mostrada abaixo -
>>> from Bio.Cluster import somcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid,map = somcluster(data)
>>> print(map)
[[[-1.36032469 0.38667395]]
[[-0.41170578 1.35295911]]]
>>> print(clusterid)
[[1 0]
[1 0]
[1 0]]Aqui, clusterid é uma matriz com duas colunas, onde o número de linhas é igual ao número de itens que foram agrupados, e data é uma matriz com dimensões de linhas ou colunas.
Análise do componente principal
A análise de componentes principais é útil para visualizar dados de alta dimensão. É um método que usa operações de matriz simples de álgebra linear e estatísticas para calcular uma projeção dos dados originais no mesmo número ou menos dimensões.
Análise de componente principal retorna uma média de coluna de tupla, coordenadas, componentes e valores próprios. Vejamos os fundamentos desse conceito.
>>> from numpy import array
>>> from numpy import mean
>>> from numpy import cov
>>> from numpy.linalg import eig
# define a matrix
>>> A = array([[1, 2], [3, 4], [5, 6]])
>>> print(A)
[[1 2]
[3 4]
[5 6]]
# calculate the mean of each column
>>> M = mean(A.T, axis = 1)
>>> print(M)
[ 3. 4.]
# center columns by subtracting column means
>>> C = A - M
>>> print(C)
[[-2. -2.]
[ 0. 0.]
[ 2. 2.]]
# calculate covariance matrix of centered matrix
>>> V = cov(C.T)
>>> print(V)
[[ 4. 4.]
[ 4. 4.]]
# eigendecomposition of covariance matrix
>>> values, vectors = eig(V)
>>> print(vectors)
[[ 0.70710678 -0.70710678]
[ 0.70710678 0.70710678]]
>>> print(values)
[ 8. 0.]Vamos aplicar os mesmos dados de matriz retangular ao módulo Bio.Cluster conforme definido abaixo -
>>> from Bio.Cluster import pca
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> columnmean, coordinates, components, eigenvalues = pca(data)
>>> print(columnmean)
[ 3. 4.]
>>> print(coordinates)
[[-2.82842712 0. ]
[ 0. 0. ]
[ 2.82842712 0. ]]
>>> print(components)
[[ 0.70710678 0.70710678]
[ 0.70710678 -0.70710678]]
>>> print(eigenvalues)
[ 4. 0.]A bioinformática é uma excelente área para aplicar algoritmos de aprendizado de máquina. Aqui, temos informações genéticas de grande número de organismos e não é possível analisar manualmente todas essas informações. Se o algoritmo de aprendizado de máquina adequado for usado, podemos extrair muitas informações úteis desses dados. Biopython fornece um conjunto útil de algoritmos para fazer o aprendizado de máquina supervisionado.
A aprendizagem supervisionada é baseada na variável de entrada (X) e na variável de saída (Y). Ele usa um algoritmo para aprender a função de mapeamento da entrada para a saída. É definido abaixo -
Y = f(X)O principal objetivo desta abordagem é aproximar a função de mapeamento e quando você tiver novos dados de entrada (x), você pode prever as variáveis de saída (Y) para esses dados.
Modelo de Regressão Logística
A regressão logística é um algoritmo de aprendizado de máquina supervisionado. É usado para descobrir a diferença entre as classes K usando a soma ponderada de variáveis preditoras. Ele calcula a probabilidade de ocorrência de um evento e pode ser usado para detecção de câncer.
Biopython fornece módulo Bio.LogisticRegression para prever variáveis com base no algoritmo de regressão logística. Atualmente, Biopython implementa algoritmo de regressão logística para apenas duas classes (K = 2).
k-vizinhos mais próximos
Os vizinhos k-mais próximos também são um algoritmo de aprendizado de máquina supervisionado. Funciona categorizando os dados com base nos vizinhos mais próximos. Biopython fornece o módulo Bio.KNN para prever variáveis com base no algoritmo de vizinhos k-mais próximos.
Baías ingénuas
Os classificadores Naive Bayes são uma coleção de algoritmos de classificação baseados no Teorema de Bayes. Não é um único algoritmo, mas uma família de algoritmos onde todos eles compartilham um princípio comum, ou seja, cada par de recursos sendo classificados é independente um do outro. Biopython fornece módulo Bio.NaiveBayes para trabalhar com o algoritmo Naive Bayes.
Modelo Markov
Um modelo de Markov é um sistema matemático definido como uma coleção de variáveis aleatórias, que experimenta a transição de um estado para outro de acordo com certas regras probabilísticas. Biopython forneceBio.MarkovModel and Bio.HMM.MarkovModel modules to work with Markov models.
Biopython possui um extenso script de teste para testar o software em diferentes condições para garantir que o software esteja livre de erros. Para executar o script de teste, baixe o código-fonte do Biopython e execute o comando abaixo -
python run_tests.pyIsso executará todos os scripts de teste e fornecerá a seguinte saída -
Python version: 2.7.12 (v2.7.12:d33e0cf91556, Jun 26 2016, 12:10:39)
[GCC 4.2.1 (Apple Inc. build 5666) (dot 3)]
Operating system: posix darwin
test_Ace ... ok
test_Affy ... ok
test_AlignIO ... ok
test_AlignIO_ClustalIO ... ok
test_AlignIO_EmbossIO ... ok
test_AlignIO_FastaIO ... ok
test_AlignIO_MauveIO ... ok
test_AlignIO_PhylipIO ... ok
test_AlignIO_convert ... ok
...........................................
...........................................Também podemos executar o script de teste individual conforme especificado abaixo -
python test_AlignIO.pyConclusão
Como vimos, Biopython é um dos softwares importantes no campo da bioinformática. Por ser escrito em python (fácil de aprender e escrever), fornece ampla funcionalidade para lidar com qualquer computação e operação no campo da bioinformática. Ele também fornece uma interface fácil e flexível para quase todos os softwares de bioinformática populares para explorar sua funcionalidade.