Biopython - Hızlı Kılavuz
Biopython, Python için en büyük ve en popüler biyoinformatik paketidir. Genel biyoinformatik görevleri için bir dizi farklı alt modül içerir. Chapman ve Chang tarafından geliştirilmiştir, çoğunlukla Python ile yazılmıştır. Ayrıca, yazılımın karmaşık hesaplama bölümünü optimize etmek için C kodu içerir. Windows, Linux, Mac OS X, vb. Üzerinde çalışır.
Temel olarak Biopython, bir DNA dizgisinin ters tamamlanması, protein dizilerinde motiflerin bulunması, vb. Gibi DNA, RNA ve protein dizisi işlemleriyle başa çıkmak için işlevler sağlayan bir python modülleri koleksiyonudur. GenBank, SwissPort, FASTA vb. gibi, python ortamında NCBI BLASTN, Entrez vb. gibi diğer popüler biyoinformatik yazılımları / araçları çalıştırmak için sarmalayıcılar / arayüzler. BioPerl, BioJava ve BioRuby gibi kardeş projeleri var.
Özellikleri
Biopython taşınabilir, net ve öğrenmesi kolay sözdizimine sahiptir. Göze çarpan özelliklerden bazıları aşağıda listelenmiştir -
Yorumlanmış, etkileşimli ve nesne yönelimli.
FASTA, PDB, GenBank, Blast, SCOP, PubMed / Medline, ExPASy ile ilgili formatları destekler.
Dizi formatlarıyla başa çıkma seçeneği.
Protein yapılarını yönetmek için araçlar.
BioSQL - Sıraları, özellikleri ve açıklamaları depolamak için standart SQL tabloları seti.
NCBI hizmetleri (Blast, Entrez, PubMed) ve ExPASY hizmetleri (SwissProt, Prosite) dahil olmak üzere çevrimiçi hizmetlere ve veritabanına erişim.
Blast, Clustalw, EMBOSS dahil olmak üzere yerel hizmetlere erişim.
Hedefler
Biopython'un amacı, python dili aracılığıyla biyoinformatiğe basit, standart ve kapsamlı erişim sağlamaktır. Biopython'un özel hedefleri aşağıda listelenmiştir -
Biyoinformatik kaynaklara standartlaştırılmış erişim sağlamak.
Yüksek kaliteli, yeniden kullanılabilir modüller ve komut dosyaları.
Küme kodu, PDB, NaiveBayes ve Markov Modelinde kullanılabilen hızlı dizi manipülasyonu.
Genomik veri analizi.
Avantajlar
Biopython çok daha az kod gerektirir ve aşağıdaki avantajları sağlar -
Kümelemede kullanılan mikrodizi veri türünü sağlar.
Ağaç Görünümü tipi dosyaları okur ve yazar.
PDB ayrıştırma, temsil ve analiz için kullanılan yapı verilerini destekler.
Medline uygulamalarında kullanılan günlük verilerini destekler.
Tüm biyoinformatik projeleri arasında yaygın olarak kullanılan standart veritabanı olan BioSQL veritabanını destekler.
Bir biyoinformatik dosyasını formata özgü bir kayıt nesnesine veya genel bir sıra sınıfı artı özelliklere ayrıştırmak için modüller sağlayarak ayrıştırıcı geliştirmeyi destekler .
Yemek kitabı stiline göre açık belgeler.
Örnek Vaka Çalışması
Bazı kullanım durumlarını (popülasyon genetiği, RNA yapısı vb.) Kontrol edelim ve Biopython'un bu alanda nasıl önemli bir rol oynadığını anlamaya çalışalım -
Popülasyon genetiği
Popülasyon genetiği, bir popülasyondaki genetik varyasyon çalışmasıdır ve popülasyonlardaki genlerin ve alellerin frekanslarındaki değişikliklerin uzay ve zaman boyunca incelenmesini ve modellenmesini içerir.
Biopython, popülasyon genetiği için Bio.PopGen modülü sağlar. Bu modül, klasik popülasyon genetiği hakkında bilgi toplamak için gerekli tüm fonksiyonları içerir.
RNA Yapısı
Yaşamımız için gerekli olan üç ana biyolojik makromolekül DNA, RNA ve Proteindir. Proteinler hücrenin yük beygisidir ve enzimler olarak önemli bir rol oynarlar. DNA (deoksiribonükleik asit) hücrenin "planı" olarak kabul edilir. Hücrenin büyümesi, besinleri alması ve çoğalması için gerekli tüm genetik bilgiyi taşır. RNA (Ribonükleik asit), hücrede "DNA fotokopisi" görevi görür.
Biopython, DNA ve RNA'nın yapı taşları olan nükleotidleri temsil eden Bio.Sequence nesnelerini sağlar.
Bu bölüm, makinenize Biopython'u nasıl kuracağınızı açıklar. Kurulumu çok kolaydır ve beş dakikadan fazla sürmez.
Step 1 - Python Kurulumunu Doğrulama
Biopython, Python 2.5 veya daha yüksek sürümlerle çalışmak üzere tasarlanmıştır. Bu nedenle, önce python'un yüklenmesi zorunludur. Komut isteminizde aşağıdaki komutu çalıştırın -
> python --versionAşağıda tanımlanmıştır -
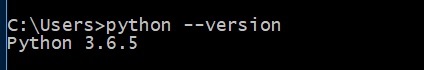
Düzgün kurulursa python sürümünü gösterir. Aksi takdirde, python'un en son sürümünü indirin, kurun ve ardından komutu yeniden çalıştırın.
Step 2 - Biopython'u pip kullanarak kurma
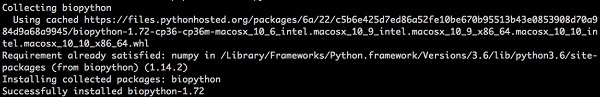
Biopython'u tüm platformlarda komut satırından pip kullanarak kurmak kolaydır. Aşağıdaki komutu yazın -
> pip install biopythonAşağıdaki yanıt ekranınızda görülecektir -
Biopython'un eski bir sürümünü güncellemek için -
> pip install biopython –-upgradeAşağıdaki yanıt ekranınızda görülecektir -
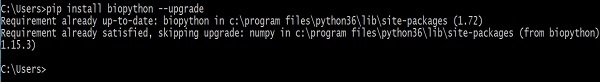
Bu komutu çalıştırdıktan sonra, son sürümleri yüklemeden önce Biopython ve NumPy'nin eski sürümleri (Biopython buna bağlıdır) kaldırılacaktır.
Step 3 - Biopython Kurulumunu Doğrulama
Şimdi, Biopython'u makinenize başarıyla yüklediniz. Biopython'un düzgün bir şekilde kurulduğunu doğrulamak için python konsolunuzda aşağıdaki komutu yazın -
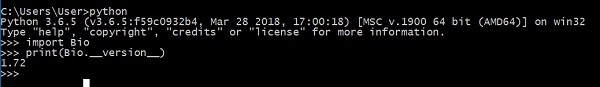
Biopython sürümünü gösterir.
Alternate Way − Installing Biopython using Source
Biopython'u kaynak kodunu kullanarak kurmak için aşağıdaki talimatları izleyin -
Aşağıdaki bağlantıdan Biopython'un son sürümünü indirin - https://biopython.org/wiki/Download
Şu an itibariyle en son sürüm biopython-1.72.
Dosyayı indirin ve sıkıştırılmış arşiv dosyasını açın, kaynak kod klasörüne gidin ve aşağıdaki komutu yazın -
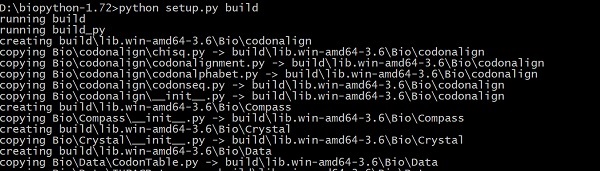
> python setup.py buildBu, Biopython'u aşağıda verildiği gibi kaynak kodundan oluşturacaktır -
Şimdi, aşağıdaki komutu kullanarak kodu test edin -
> python setup.py test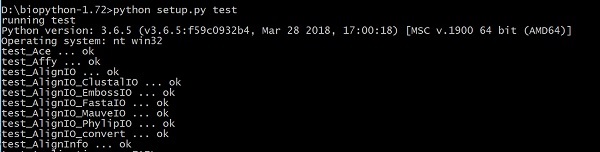
Son olarak, aşağıdaki komutu kullanarak kurun -
> python setup.py install
Bir biyoinformatik dosyasını ayrıştırmak ve içeriği yazdırmak için basit bir Biopython uygulaması oluşturalım. Bu, Biopython'un genel konseptini ve biyoinformatik alanında nasıl yardımcı olduğunu anlamamıza yardımcı olacaktır.
Step 1 - Önce örnek bir sıra dosyası oluşturun, "example.fasta" ve aşağıdaki içeriği içine koyun.
>sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAV
NNFEAHTINTVVHTNDSDKGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITID
SNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTAGQYQGLVSIILTKSTTTTTTTKGT
>sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVS
NTLVGVLTLSNTSIDTVSIASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDK
NAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGNYRANITITSTIKGGGTKKGTTDKKFasta uzantısı, sıra dosyasının dosya biçimini ifade eder. FAŞTA, biyoinformatik yazılımı olan FAŞTA'dan geliyor ve bu nedenle adını alıyor. FAŞTA formatı, tek tek düzenlenmiş birden fazla diziye sahiptir ve her dizinin kendi kimliği, adı, açıklaması ve gerçek dizi verileri olacaktır.
Step 2 - Yeni bir python betiği oluşturun, * simple_example.py "ve aşağıdaki kodu girin ve kaydedin.
from Bio.SeqIO import parse
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
file = open("example.fasta")
records = parse(file, "fasta") for record in records:
print("Id: %s" % record.id)
print("Name: %s" % record.name)
print("Description: %s" % record.description)
print("Annotations: %s" % record.annotations)
print("Sequence Data: %s" % record.seq)
print("Sequence Alphabet: %s" % record.seq.alphabet)Kodu biraz daha derinlemesine inceleyelim -
Line 1Bio.SeqIO modülünde bulunan ayrıştırma sınıfını içe aktarır. Bio.SeqIO modülü, sekans dosyasını farklı formatta okumak ve yazmak için kullanılır ve sekans dosyasının içeriğini ayrıştırmak için `` ayrıştırma '' sınıfı kullanılır.
Line 2Bio.SeqRecord modülünde bulunan SeqRecord sınıfını içe aktarır. Bu modül, sekans kayıtlarını işlemek için kullanılır ve SeqRecord sınıfı, sekans dosyasında bulunan belirli bir sekansı temsil etmek için kullanılır.
*Line 3"Bio.Seq modülünde bulunan Seq sınıfını içe aktarır. Bu modül, sekans verilerini işlemek için kullanılır ve Seq sınıfı, sekans dosyasında bulunan belirli bir sekans kaydının sekans verilerini temsil etmek için kullanılır.
Line 5 normal python işlevini kullanarak “example.fasta” dosyasını açar, açın.
Line 7 sıra dosyasının içeriğini ayrıştırır ve içeriği SeqRecord nesnesinin listesi olarak döndürür.
Line 9-15 python for döngüsü kullanarak kayıtlar üzerinde döngü oluşturur ve id, ad, açıklama, sıra verileri vb. gibi sıralama kaydının (SqlRecord) niteliklerini yazdırır.
Line 15 Alfabe sınıfını kullanarak dizinin türünü yazdırır.
Step 3 - Bir komut istemi açın ve "example.fasta" adlı sıra dosyasını içeren klasöre gidin ve aşağıdaki komutu çalıştırın -
> python simple_example.pyStep 4- Python, komut dosyasını çalıştırır ve örnek dosyada bulunan tüm sıra verilerini ("example.fasta") yazdırır. Çıktı aşağıdaki içeriğe benzer olacaktır.
Id: sp|P25730|FMS1_ECOLI
Name: sp|P25730|FMS1_ECOLI
Decription: sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
Annotations: {}
Sequence Data: MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAVNNFEAHTINTVVHTNDSD
KGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITIDSNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTA
GQYQGLVSIILTKSTTTTTTTKGT
Sequence Alphabet: SingleLetterAlphabet()
Id: sp|P15488|FMS3_ECOLI
Name: sp|P15488|FMS3_ECOLI
Decription: sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
Annotations: {}
Sequence Data: MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVSNTLVGVLTLSNTSIDTVS
IASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDKNAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGN
YRANITITSTIKGGGTKKGTTDKK
Sequence Alphabet: SingleLetterAlphabet()Bu örnekte ayrıştırma, SeqRecord ve Seq olmak üzere üç sınıf gördük. Bu üç sınıf, işlevselliğin çoğunu sağlar ve bu sınıfları ilerleyen bölümde öğreneceğiz.
Bir dizi, bir organizmanın proteinini, DNA'sını veya RNA'sını temsil etmek için kullanılan bir dizi harftir. Seq sınıfı ile temsil edilir. Seq sınıfı Bio.Seq modülünde tanımlanmıştır.
Biopython'da aşağıda gösterildiği gibi basit bir dizi oluşturalım -
>>> from Bio.Seq import Seq
>>> seq = Seq("AGCT")
>>> seq
Seq('AGCT')
>>> print(seq)
AGCTBurada basit bir protein dizisi oluşturduk AGCT ve her harf temsil eder Aşerit Glikin Cysteine ve Threonin.
Her Seq nesnesinin iki önemli özelliği vardır -
veri - gerçek sıra dizisi (AGCT)
alfabe - dizinin türünü temsil etmek için kullanılır. örneğin, DNA dizisi, RNA dizisi, vb. Varsayılan olarak, herhangi bir diziyi temsil etmez ve doğası gereği jeneriktir.
Alfabe Modülü
Seq nesneleri, sıra türünü, harfleri ve olası işlemleri belirtmek için Alfabe niteliği içerir. Bio.Alphabet modülünde tanımlanmıştır. Alfabe aşağıdaki gibi tanımlanabilir -
>>> from Bio.Seq import Seq
>>> myseq = Seq("AGCT")
>>> myseq
Seq('AGCT')
>>> myseq.alphabet
Alphabet()Alfabe modülü, farklı sekans türlerini temsil etmek için aşağıdaki sınıfları sağlar. Alfabe - tüm alfabe türleri için temel sınıf.
SingleLetterAlphabet - Bir boyuttaki harflere sahip genel alfabe. Alfabe'den türetilir ve diğer tüm alfabe türleri ondan türetilir.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import single_letter_alphabet
>>> test_seq = Seq('AGTACACTGGT', single_letter_alphabet)
>>> test_seq
Seq('AGTACACTGGT', SingleLetterAlphabet())ProteinAlphabet - Genel tek harfli protein alfabesi.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_protein
>>> test_seq = Seq('AGTACACTGGT', generic_protein)
>>> test_seq
Seq('AGTACACTGGT', ProteinAlphabet())NucleotideAlphabet - Genel tek harfli nükleotid alfabesi.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_nucleotide
>>> test_seq = Seq('AGTACACTGGT', generic_nucleotide) >>> test_seq
Seq('AGTACACTGGT', NucleotideAlphabet())DNAAlphabet - Genel tek harfli DNA alfabesi.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_dna
>>> test_seq = Seq('AGTACACTGGT', generic_dna)
>>> test_seq
Seq('AGTACACTGGT', DNAAlphabet())RNAAlphabet - Genel tek harfli RNA alfabesi.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_rna
>>> test_seq = Seq('AGTACACTGGT', generic_rna)
>>> test_seq
Seq('AGTACACTGGT', RNAAlphabet())Biopython modülü, Bio.Alphabet.IUPAC, IUPAC topluluğu tarafından tanımlanan temel dizi türlerini sağlar. Aşağıdaki sınıfları içerir -
IUPACProtein (protein) - 20 standart amino asitten oluşan IUPAC protein alfabesi.
ExtendedIUPACProtein (extended_protein) - X dahil olmak üzere genişletilmiş büyük IUPAC protein tek harfli alfabe.
IUPACAmbiguousDNA (ambiguous_dna) - Büyük IUPAC belirsiz DNA'sı.
IUPACUnambiguousDNA (unambiguous_dna) - Büyük IUPAC kesin DNA (GATC).
ExtendedIUPACDNA (extended_dna) - Genişletilmiş IUPAC DNA alfabesi.
IUPACAmbiguousRNA (ambiguous_rna) - Büyük harfli IUPAC belirsiz RNA.
IUPACUnambiguousRNA (unambiguous_rna) - Büyük IUPAC kesin RNA (GAUC).
Aşağıda gösterildiği gibi IUPACProtein sınıfı için basit bir örnek düşünün -
>>> from Bio.Alphabet import IUPAC
>>> protein_seq = Seq("AGCT", IUPAC.protein)
>>> protein_seq
Seq('AGCT', IUPACProtein())
>>> protein_seq.alphabetAyrıca Biopython, Bio.Data modülü aracılığıyla biyoinformatikle ilgili tüm yapılandırma verilerini ortaya çıkarır. Örneğin, IUPACData.protein_letters, IUPACProtein alfabesinin olası harflerine sahiptir.
>>> from Bio.Data import IUPACData
>>> IUPACData.protein_letters
'ACDEFGHIKLMNPQRSTVWY'Temel işlemler
Bu bölüm Seq sınıfında bulunan tüm temel işlemleri kısaca açıklamaktadır. Diziler python dizelerine benzer. Sıralı olarak dilimleme, sayma, birleştirme, bulma, ayırma ve ayırma gibi python string işlemlerini gerçekleştirebiliriz.
Çeşitli çıktılar elde etmek için aşağıdaki kodları kullanın.
To get the first value in sequence.
>>> seq_string = Seq("AGCTAGCT")
>>> seq_string[0]
'A'To print the first two values.
>>> seq_string[0:2]
Seq('AG')To print all the values.
>>> seq_string[ : ]
Seq('AGCTAGCT')To perform length and count operations.
>>> len(seq_string)
8
>>> seq_string.count('A')
2To add two sequences.
>>> from Bio.Alphabet import generic_dna, generic_protein
>>> seq1 = Seq("AGCT", generic_dna)
>>> seq2 = Seq("TCGA", generic_dna)
>>> seq1+seq2
Seq('AGCTTCGA', DNAAlphabet())Burada, yukarıdaki iki sekans nesnesi, seq1, seq2, jenerik DNA sekanslarıdır ve böylece onları ekleyebilir ve yeni sekans üretebilirsiniz. Bir protein dizisi ve aşağıda belirtildiği gibi bir DNA dizisi gibi uyumsuz alfabelere sahip diziler ekleyemezsiniz -
>>> dna_seq = Seq('AGTACACTGGT', generic_dna)
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> dna_seq + protein_seq
.....
.....
TypeError: Incompatible alphabets DNAAlphabet() and ProteinAlphabet()
>>>İki veya daha fazla dizi eklemek için önce bir python listesinde saklayın, ardından 'for döngüsü' kullanarak geri alın ve son olarak aşağıda gösterildiği gibi bir araya ekleyin -
>>> from Bio.Alphabet import generic_dna
>>> list = [Seq("AGCT",generic_dna),Seq("TCGA",generic_dna),Seq("AAA",generic_dna)]
>>> for s in list:
... print(s)
...
AGCT
TCGA
AAA
>>> final_seq = Seq(" ",generic_dna)
>>> for s in list:
... final_seq = final_seq + s
...
>>> final_seq
Seq('AGCTTCGAAAA', DNAAlphabet())Aşağıdaki bölümde ihtiyaca göre çıktı almak için çeşitli kodlar verilmiştir.
To change the case of sequence.
>>> from Bio.Alphabet import generic_rna
>>> rna = Seq("agct", generic_rna)
>>> rna.upper()
Seq('AGCT', RNAAlphabet())To check python membership and identity operator.
>>> rna = Seq("agct", generic_rna)
>>> 'a' in rna
True
>>> 'A' in rna
False
>>> rna1 = Seq("AGCT", generic_dna)
>>> rna is rna1
FalseTo find single letter or sequence of letter inside the given sequence.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.find('G')
1
>>> protein_seq.find('GG')
8To perform splitting operation.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.split('A')
[Seq('', ProteinAlphabet()), Seq('GU', ProteinAlphabet()),
Seq('C', ProteinAlphabet()), Seq('CUGGU', ProteinAlphabet())]To perform strip operations in the sequence.
>>> strip_seq = Seq(" AGCT ")
>>> strip_seq
Seq(' AGCT ')
>>> strip_seq.strip()
Seq('AGCT')Bu bölümde, Biopython tarafından sağlanan gelişmiş dizi özelliklerinden bazılarını tartışacağız.
Tamamlayıcı ve Ters Tamamlayıcı
Nükleotid sekansı, yeni sekans elde etmek için tersine tamamlanabilir. Ayrıca, tamamlanan dizi, orijinal diziyi elde etmek için ters tamamlanabilir. Biopython, bu işlevi yapmak için iki yöntem sağlar -complement ve reverse_complement. Bunun kodu aşağıda verilmiştir -
>>> from Bio.Alphabet import IUPAC
>>> nucleotide = Seq('TCGAAGTCAGTC', IUPAC.ambiguous_dna)
>>> nucleotide.complement()
Seq('AGCTTCAGTCAG', IUPACAmbiguousDNA())
>>>Burada, kompleman () yöntemi, bir DNA veya RNA dizisini tamamlamaya izin verir. Reverse_complement () yöntemi, sonuçtaki diziyi soldan sağa tamamlar ve tersine çevirir. Aşağıda gösterilmiştir -
>>> nucleotide.reverse_complement()
Seq('GACTGACTTCGA', IUPACAmbiguousDNA())Biopython, tamamlama işlemini yapmak için Bio.Data.IUPACData tarafından sağlanan ambiguous_dna_complement değişkenini kullanır.
>>> from Bio.Data import IUPACData
>>> import pprint
>>> pprint.pprint(IUPACData.ambiguous_dna_complement) {
'A': 'T',
'B': 'V',
'C': 'G',
'D': 'H',
'G': 'C',
'H': 'D',
'K': 'M',
'M': 'K',
'N': 'N',
'R': 'Y',
'S': 'S',
'T': 'A',
'V': 'B',
'W': 'W',
'X': 'X',
'Y': 'R'}
>>>GC İçeriği
Genomik DNA baz bileşiminin (GC içeriği), genom işleyişini ve tür ekolojisini önemli ölçüde etkilediği tahmin edilmektedir. GC içeriği, GC nükleotidlerinin sayısının toplam nükleotidlere bölünmesiyle elde edilir.
GC nükleotid içeriğini almak için aşağıdaki modülü içe aktarın ve aşağıdaki adımları gerçekleştirin -
>>> from Bio.SeqUtils import GC
>>> nucleotide = Seq("GACTGACTTCGA",IUPAC.unambiguous_dna)
>>> GC(nucleotide)
50.0Transkripsiyon
Transkripsiyon, DNA dizisini RNA dizisine dönüştürme işlemidir. Gerçek biyolojik transkripsiyon işlemi, DNA'yı şablon zincir olarak dikkate alarak mRNA'yı elde etmek için bir ters tamamlayıcı (TCAG → CUGA) gerçekleştiriyor. Bununla birlikte, biyoinformatikte ve dolayısıyla Biopython'da, genellikle doğrudan kodlama dizisiyle çalışırız ve mRNA dizisini T harfini U olarak değiştirerek elde edebiliriz.
Yukarıdakiler için basit bir örnek aşağıdaki gibidir -
>>> from Bio.Seq import Seq
>>> from Bio.Seq import transcribe
>>> from Bio.Alphabet import IUPAC
>>> dna_seq = Seq("ATGCCGATCGTAT",IUPAC.unambiguous_dna) >>> transcribe(dna_seq)
Seq('AUGCCGAUCGUAU', IUPACUnambiguousRNA())
>>>Transkripsiyonu tersine çevirmek için, aşağıdaki kodda gösterildiği gibi T, U olarak değiştirilir -
>>> rna_seq = transcribe(dna_seq)
>>> rna_seq.back_transcribe()
Seq('ATGCCGATCGTAT', IUPACUnambiguousDNA())DNA şablon zincirini elde etmek için, aşağıda verildiği gibi ters kopyalanmış RNA'yı ters tamamlayın -
>>> rna_seq.back_transcribe().reverse_complement()
Seq('ATACGATCGGCAT', IUPACUnambiguousDNA())Tercüme
Çeviri, RNA dizisini protein dizisine çevirme işlemidir. Aşağıda gösterildiği gibi bir RNA dizisi düşünün -
>>> rna_seq = Seq("AUGGCCAUUGUAAU",IUPAC.unambiguous_rna)
>>> rna_seq
Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())Şimdi, translate () işlevini yukarıdaki koda uygulayın -
>>> rna_seq.translate()
Seq('MAIV', IUPACProtein())Yukarıdaki RNA dizisi basittir. AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA RNA dizisini düşünün ve translate () uygulayın -
>>> rna = Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA', IUPAC.unambiguous_rna)
>>> rna.translate()
Seq('MAIVMGR*KGAR', HasStopCodon(IUPACProtein(), '*'))Burada, durdurma kodonları bir yıldız işareti '*' ile gösterilir.
Translate () yönteminde ilk durdurma kodonunda durmak mümkündür. Bunu yapmak için translate () 'de to_stop = True atayabilirsiniz:
>>> rna.translate(to_stop = True)
Seq('MAIVMGR', IUPACProtein())Burada, durdurma kodonu, bir tane içermediğinden sonuç dizisine dahil edilmez.
Çeviri Tablosu
NCBI'nin Genetik Kodlar sayfası, Biopython tarafından kullanılan çeviri tablolarının tam listesini sağlar. Kodu görselleştirmek için standart tablo için bir örnek görelim -
>>> from Bio.Data import CodonTable
>>> table = CodonTable.unambiguous_dna_by_name["Standard"]
>>> print(table)
Table 1 Standard, SGC0
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--
>>>Biopython, bu tabloyu DNA'yı proteine çevirmek ve ayrıca Durdurma kodonunu bulmak için kullanır.
Biopython, sıraları bir dosyadan (herhangi bir akış) okumak ve dosyaya yazmak için Bio.SeqIO adlı bir modül sağlar. Biyoinformatikte bulunan neredeyse tüm dosya formatlarını destekler. Yazılımın çoğu, farklı dosya formatları için farklı yaklaşımlar sunar. Ancak Biopython, ayrıştırılmış dizi verilerini SeqRecord nesnesi aracılığıyla kullanıcıya sunmak için bilinçli olarak tek bir yaklaşımı izler.
Aşağıdaki bölümde SeqRecord hakkında daha fazla bilgi edinelim.
SeqRecord
Bio.SeqRecord modülü, SeqRecord'un aşağıda verildiği gibi sekansın meta bilgilerini ve sekans verilerini tutmasını sağlar -
seq - Bu gerçek bir dizidir.
id - Verilen dizinin birincil tanımlayıcısıdır. Varsayılan tür dizedir.
isim - Sıranın Adıdır. Varsayılan tür dizedir.
açıklama - Dizi hakkında okunabilir bilgileri görüntüler.
ek açıklamalar - Sırayla ilgili ek bilgilerin bulunduğu bir sözlüktür.
SeqRecord aşağıda belirtildiği gibi içe aktarılabilir
from Bio.SeqRecord import SeqRecordİlerleyen bölümlerde sıra dosyasını gerçek sıra dosyası kullanarak ayrıştırmanın nüanslarını anlayalım.
Sıralı Dosya Biçimlerini Ayrıştırma
Bu bölümde, en popüler iki dizi dosya biçiminin nasıl ayrıştırılacağı açıklanmaktadır. FASTA ve GenBank.
FAŞTA
FASTAsıra verilerini depolamak için en temel dosya formatıdır. Başlangıçta FAŞTA, Biyoinformatiğin erken evrimi sırasında geliştirilen ve çoğunlukla dizi benzerliğini araştırmak için kullanılan DNA ve proteinin dizi hizalaması için bir yazılım paketidir.
Biopython, örnek bir FASTA dosyası sağlar ve şu adresten erişilebilir: https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Bu dosyayı indirin ve Biopython örnek dizininize şu şekilde kaydedin: ‘orchid.fasta’.
Bio.SeqIO modülü, sıra dosyalarını işlemek için parse () yöntemi sağlar ve aşağıdaki şekilde içe aktarılabilir -
from Bio.SeqIO import parseparse () yöntemi iki bağımsız değişken içerir, birincisi dosya tanıtıcısı ve ikincisi dosya biçimidir.
>>> file = open('path/to/biopython/sample/orchid.fasta')
>>> for record in parse(file, "fasta"):
... print(record.id)
...
gi|2765658|emb|Z78533.1|CIZ78533
gi|2765657|emb|Z78532.1|CCZ78532
..........
..........
gi|2765565|emb|Z78440.1|PPZ78440
gi|2765564|emb|Z78439.1|PBZ78439
>>>Burada, parse () yöntemi her yinelemede SeqRecord döndüren yinelenebilir bir nesne döndürür. Tekrarlanabilir olması, birçok karmaşık ve kolay yöntem sağlar ve bazı özellikleri görmemize izin verir.
Sonraki()
next () yöntemi, aşağıda verildiği gibi ilk diziyi elde etmek için kullanabileceğimiz, yinelenebilir nesnede bulunan sonraki öğeyi döndürür -
>>> first_seq_record = next(SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta'))
>>> first_seq_record.id 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.name 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', SingleLetterAlphabet())
>>> first_seq_record.description 'gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> first_seq_record.annotations
{}
>>>Burada seq_record.annotations boştur çünkü FAŞTA formatı sıra açıklamalarını desteklemez.
liste anlama
Yinelenebilir nesneyi, aşağıda verilen liste kavrayışını kullanarak listeye dönüştürebiliriz.
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> all_seq = [seq_record for seq_record in seq_iter] >>> len(all_seq)
94
>>>Burada toplam sayımı elde etmek için len yöntemini kullandık. Maksimum uzunluktaki diziyi aşağıdaki gibi alabiliriz -
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> max_seq = max(len(seq_record.seq) for seq_record in seq_iter)
>>> max_seq
789
>>>Aşağıdaki kodu kullanarak sıralamayı da filtreleyebiliriz -
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> seq_under_600 = [seq_record for seq_record in seq_iter if len(seq_record.seq) < 600]
>>> for seq in seq_under_600:
... print(seq.id)
...
gi|2765606|emb|Z78481.1|PIZ78481
gi|2765605|emb|Z78480.1|PGZ78480
gi|2765601|emb|Z78476.1|PGZ78476
gi|2765595|emb|Z78470.1|PPZ78470
gi|2765594|emb|Z78469.1|PHZ78469
gi|2765564|emb|Z78439.1|PBZ78439
>>>Bir SqlRecord nesneleri koleksiyonunu (ayrıştırılmış veriler) dosyaya yazmak, aşağıdaki gibi SeqIO.write yöntemini çağırmak kadar basittir -
file = open("converted.fasta", "w)
SeqIO.write(seq_record, file, "fasta")Bu yöntem, biçimi aşağıda belirtildiği gibi dönüştürmek için etkili bir şekilde kullanılabilir -
file = open("converted.gbk", "w)
SeqIO.write(seq_record, file, "genbank")GenBank
Genler için daha zengin bir dizi formatıdır ve çeşitli ek açıklama türleri için alanlar içerir. Biopython, örnek bir GenBank dosyası sağlar ve şu adresten erişilebilir:https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Dosyayı Biopython örnek dizininize indirin ve kaydedin. ‘orchid.gbk’
Biopython tek bir işlev sağladığından, tüm biyoinformatik formatını ayrıştırmak için ayrıştırın. GenBank formatının ayrıştırılması, ayrıştırma yöntemindeki format seçeneğini değiştirmek kadar basittir.
Aynı kod aşağıda verilmiştir -
>>> from Bio import SeqIO
>>> from Bio.SeqIO import parse
>>> seq_record = next(parse(open('path/to/biopython/sample/orchid.gbk'),'genbank'))
>>> seq_record.id
'Z78533.1'
>>> seq_record.name
'Z78533'
>>> seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', IUPACAmbiguousDNA())
>>> seq_record.description
'C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> seq_record.annotations {
'molecule_type': 'DNA',
'topology': 'linear',
'data_file_division': 'PLN',
'date': '30-NOV-2006',
'accessions': ['Z78533'],
'sequence_version': 1,
'gi': '2765658',
'keywords': ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'],
'source': 'Cypripedium irapeanum',
'organism': 'Cypripedium irapeanum',
'taxonomy': [
'Eukaryota',
'Viridiplantae',
'Streptophyta',
'Embryophyta',
'Tracheophyta',
'Spermatophyta',
'Magnoliophyta',
'Liliopsida',
'Asparagales',
'Orchidaceae',
'Cypripedioideae',
'Cypripedium'],
'references': [
Reference(title = 'Phylogenetics of the slipper orchids (Cypripedioideae:
Orchidaceae): nuclear rDNA ITS sequences', ...),
Reference(title = 'Direct Submission', ...)
]
}Sequence alignment aralarındaki benzerlik bölgesini belirlemek için iki veya daha fazla diziyi (DNA, RNA veya protein dizilerinin) belirli bir sırayla düzenleme işlemidir.
Benzer bölgeyi belirlemek, türler arasında hangi özelliklerin korunduğu, farklı türlerin genetik olarak ne kadar yakın olduğu, türlerin nasıl evrimleştiği vb. Gibi birçok bilgiyi çıkarmamızı sağlar. Biopython, dizi hizalaması için kapsamlı destek sağlar.
Bu bölümde Biopython tarafından sağlanan bazı önemli özellikleri öğrenelim -
Ayrıştırma Sırası Hizalaması
Biopython, sıra hizalamalarını okumak ve yazmak için Bio.AlignIO adlı bir modül sağlar. Biyoinformatikte, daha önce öğrenilen dizi verilerine benzer dizi hizalama verilerini belirtmek için birçok format mevcuttur. Bio.AlignIO, Bio.SeqIO'nun sekans verileri üzerinde çalışması ve Bio.AlignIO'nun sekans hizalama verileri üzerinde çalışması dışında Bio.SeqIO'ya benzer API sağlar.
Öğrenmeye başlamadan önce, internetten örnek bir sıra hizalama dosyası indirelim.
Örnek dosyayı indirmek için aşağıdaki adımları izleyin -
Step 1 - Favori tarayıcınızı açın ve şu adrese gidin: http://pfam.xfam.org/family/browseİnternet sitesi. Tüm Pfam ailelerini alfabetik sırayla gösterecektir.
Step 2- Daha az sayıda tohum değerine sahip herhangi bir aile seçin. Minimum veri içerir ve hizalamayla kolayca çalışmamızı sağlar. Burada, PF18225'i seçtik / tıkladık ve açılırhttp://pfam.xfam.org/family/PF18225 ve sıra hizalamaları da dahil olmak üzere bununla ilgili tüm ayrıntıları gösterir.
Step 3 - Hizalama bölümüne gidin ve sıra hizalama dosyasını Stockholm formatında indirin (PF18225_seed.txt).
Bio.AlignIO kullanarak indirilen sıralama hizalama dosyasını aşağıdaki gibi okumaya çalışalım -
Bio.AlignIO modülünü içe aktar
>>> from Bio import AlignIOOkuma yöntemini kullanarak hizalamayı okuyun. okuma yöntemi, verilen dosyada bulunan tek hizalama verilerini okumak için kullanılır. Verilen dosya çok sayıda hizalama içeriyorsa, ayrıştırma yöntemini kullanabiliriz. ayrıştırma yöntemi Bio.SeqIO modülündeki ayrıştırma yöntemine benzer yinelenebilir hizalama nesnesi döndürür.
>>> alignment = AlignIO.read(open("PF18225_seed.txt"), "stockholm")Hizalama nesnesini yazdırın.
>>> print(alignment)
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Ayrıca hizalamada bulunan dizileri (SeqRecord) ve aşağıda kontrol edebiliriz -
>>> for align in alignment:
... print(align.seq)
...
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVATVANQLRGRKRRAFARHREGP
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADITA---RLDRRREHGEHGVRKKP
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMAPMLIALNYRNRESHAQVDKKP
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMAPLFKVLSFRNREDQGLVNNKP
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIMVLAPRLTAKHPYDKVQDRNRK
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVADLMRKLDLDRPFKKLERKNRT
>>>Çoklu Hizalama
Genel olarak, sıra hizalama dosyalarının çoğu tek hizalama verisi içerir ve kullanılması yeterlidir readayrıştırma yöntemi. Çoklu dizi hizalama konseptinde, iki veya daha fazla dizi, aralarındaki en iyi alt dizi eşleşmeleri için karşılaştırılır ve tek bir dosyada çoklu dizi hizalamasına neden olur.
Giriş dizisi hizalama biçimi birden fazla dizi hizalaması içeriyorsa, o zaman kullanmamız gerekir parse yerine yöntem read aşağıda belirtildiği gibi yöntem -
>>> from Bio import AlignIO
>>> alignments = AlignIO.parse(open("PF18225_seed.txt"), "stockholm")
>>> print(alignments)
<generator object parse at 0x000001CD1C7E0360>
>>> for alignment in alignments:
... print(alignment)
...
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Burada, ayrıştırma yöntemi yinelenebilir hizalama nesnesi döndürür ve gerçek hizalamaları elde etmek için yinelenebilir.
İkili Sıra Hizalama
Pairwise sequence alignment aynı anda yalnızca iki diziyi karşılaştırır ve olası en iyi dizi hizalamalarını sağlar. Pairwise anlaşılması kolaydır ve ortaya çıkan dizi hizalamasından çıkarılması olağanüstüdür.
Biopython özel bir modül sağlar, Bio.pairwise2ikili yöntemi kullanarak hizalama sırasını tanımlamak için. Biopython, hizalama sırasını bulmak için en iyi algoritmayı uygular ve diğer yazılımlarla aynıdır.
İkili modül kullanarak iki basit ve varsayımsal dizinin dizi hizalamasını bulmak için bir örnek yazalım. Bu, dizi hizalama kavramını ve bunu Biopython kullanarak nasıl programlayacağımızı anlamamıza yardımcı olacaktır.
Aşama 1
Modülü içe aktarın pairwise2 aşağıda verilen komutla -
>>> from Bio import pairwise2Adım 2
Seq1 ve seq2 olmak üzere iki dizi oluşturun -
>>> from Bio.Seq import Seq
>>> seq1 = Seq("ACCGGT")
>>> seq2 = Seq("ACGT")Aşama 3
Aşağıdaki kod satırını kullanarak hizalamaları bulmak için seq1 ve seq2 ile birlikte pairwise2.align.globalxx yöntemini çağırın -
>>> alignments = pairwise2.align.globalxx(seq1, seq2)Buraya, globalxxyöntem, fiili çalışmayı gerçekleştirir ve verilen dizilerde mümkün olan en iyi hizalamaları bulur. Aslında, Bio.pairwise2, farklı senaryolarda hizalamaları bulmak için aşağıdaki kuralı izleyen bir dizi yöntem sağlar.
<sequence alignment type>XYBurada, sıra hizalama türü, genel veya yerel olabilen hizalama türünü ifade eder . global tip, tüm diziyi dikkate alarak dizi hizalamasını bulmaktır. yerel tip, verilen dizilerin alt kümesine de bakarak dizi hizalamasını bulmaktır. Bu sıkıcı olacaktır, ancak verilen diziler arasındaki benzerlik hakkında daha iyi fikir verir.
X, eşleşen puanı ifade eder. Olası değerler, x (tam eşleşme), m (aynı karakterlere dayalı puan), d (karakter ve eşleşme puanına sahip kullanıcı tarafından sağlanan sözlük) ve son olarak c (özel puanlama algoritması sağlamak için kullanıcı tanımlı işlev) şeklindedir.
Y boşluk cezasını ifade eder. Olası değerler, x (boşluk cezası yok), s (her iki sıra için aynı cezalar), d (her sıra için farklı cezalar) ve son olarak c (özel boşluk cezaları sağlamak için kullanıcı tanımlı işlev)
Dolayısıyla, localds, yerel hizalama tekniğini, kullanıcı tarafından sağlanan eşleşmeler için sözlüğü ve her iki dizi için kullanıcı tarafından sağlanan boşluk cezasını kullanarak dizi hizalamasını bulan geçerli bir yöntemdir.
>>> test_alignments = pairwise2.align.localds(seq1, seq2, blosum62, -10, -1)Burada, blosum62, maç skoru sağlamak için pairwise2 modülünde bulunan bir sözlüğü ifade eder. -10 boşluk açma cezasını ve -1 boşluk uzatma cezasını ifade eder.
4. adım
Yinelenebilir hizalama nesnesi üzerinde döngü yapın ve her bir ayrı hizalama nesnesini alıp yazdırın.
>>> for alignment in alignments:
... print(alignment)
...
('ACCGGT', 'A-C-GT', 4.0, 0, 6)
('ACCGGT', 'AC--GT', 4.0, 0, 6)
('ACCGGT', 'A-CG-T', 4.0, 0, 6)
('ACCGGT', 'AC-G-T', 4.0, 0, 6)Adım 5
Bio.pairwise2 modülü, sonucu daha iyi görselleştirmek için bir formatlama yöntemi, format_alignment sağlar -
>>> from Bio.pairwise2 import format_alignment
>>> alignments = pairwise2.align.globalxx(seq1, seq2)
>>> for alignment in alignments:
... print(format_alignment(*alignment))
...
ACCGGT
| | ||
A-C-GT
Score=4
ACCGGT
|| ||
AC--GT
Score=4
ACCGGT
| || |
A-CG-T
Score=4
ACCGGT
|| | |
AC-G-T
Score=4
>>>Biopython ayrıca sıra hizalaması yapmak için başka bir modül olan Align sağlar. Bu modül, algoritma, mod, maç skoru, boşluk cezaları, vb. Gibi parametrelerin ayarlanması için farklı bir API seti sağlar. Align nesnesine basit bir bakış aşağıdaki gibidir -
>>> from Bio import Align
>>> aligner = Align.PairwiseAligner()
>>> print(aligner)
Pairwise sequence aligner with parameters
match score: 1.000000
mismatch score: 0.000000
target open gap score: 0.000000
target extend gap score: 0.000000
target left open gap score: 0.000000
target left extend gap score: 0.000000
target right open gap score: 0.000000
target right extend gap score: 0.000000
query open gap score: 0.000000
query extend gap score: 0.000000
query left open gap score: 0.000000
query left extend gap score: 0.000000
query right open gap score: 0.000000
query right extend gap score: 0.000000
mode: global
>>>Sıra Hizalama Araçları Desteği
Biopython, Bio.Align.Applications modülü aracılığıyla birçok dizi hizalama aracına arayüz sağlar. Araçlardan bazıları aşağıda listelenmiştir -
- ClustalW
- MUSCLE
- EMBOSS iğne ve su
En popüler hizalama aracı olan ClustalW ile dizi hizalaması oluşturmak için Biopython'da basit bir örnek yazalım.
Step 1 - Clustalw programını şuradan indirin: http://www.clustal.org/download/current/ve kurun. Ayrıca, PATH sistem yolunu "clustal" kurulum yolu ile güncelleyin.
Step 2 - Bio.Align.Applications modülünden ClustalwCommanLine'ı içe aktarın.
>>> from Bio.Align.Applications import ClustalwCommandlineStep 3 - Girdi dosyasıyla ClustalwCommanLine'ı çağırarak cmd'yi ayarlayın, opuntia.fasta Biopython paketinde mevcuttur. https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/opuntia.fasta
>>> cmd = ClustalwCommandline("clustalw2",
infile="/path/to/biopython/sample/opuntia.fasta")
>>> print(cmd)
clustalw2 -infile=fasta/opuntia.fastaStep 4 - cmd () 'yi çağırmak clustalw komutunu çalıştıracak ve sonuçta ortaya çıkan hizalama dosyasının opuntia.aln çıktısını verecektir.
>>> stdout, stderr = cmd()Step 5 - Hizalama dosyasını aşağıdaki gibi okuyun ve yazdırın -
>>> from Bio import AlignIO
>>> align = AlignIO.read("/path/to/biopython/sample/opuntia.aln", "clustal")
>>> print(align)
SingleLetterAlphabet() alignment with 7 rows and 906 columns
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273285|gb|AF191659.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273284|gb|AF191658.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273287|gb|AF191661.1|AF191
TATACATAAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273286|gb|AF191660.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273290|gb|AF191664.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273289|gb|AF191663.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273291|gb|AF191665.1|AF191
>>>BLAST, şu anlama gelir: Basic Local Alignment Search Tool. Biyolojik diziler arasındaki benzerlik bölgelerini bulur. Biopython, NCBI BLAST işlemi ile başa çıkmak için Bio.Blast modülü sağlar. BLAST'ı yerel bağlantıda veya İnternet bağlantısı üzerinden çalıştırabilirsiniz.
Aşağıdaki bölümde bu iki bağlantıyı kısaca anlayalım -
İnternet üzerinden çalışıyor
Biopython, BLAST'ın çevrimiçi sürümünü aramak için Bio.Blast.NCBIWWW modülü sağlar. Bunu yapmak için aşağıdaki modülü içe aktarmamız gerekiyor -
>>> from Bio.Blast import NCBIWWWNCBIWW modülü, BLAST çevrimiçi sürümünü sorgulamak için qblast işlevi sağlar, https://blast.ncbi.nlm.nih.gov/Blast.cgi. qblast, çevrimiçi sürüm tarafından desteklenen tüm parametreleri destekler.
Bu modül hakkında herhangi bir yardım almak için aşağıdaki komutu kullanın ve özellikleri anlayın -
>>> help(NCBIWWW.qblast)
Help on function qblast in module Bio.Blast.NCBIWWW:
qblast(
program, database, sequence,
url_base = 'https://blast.ncbi.nlm.nih.gov/Blast.cgi',
auto_format = None,
composition_based_statistics = None,
db_genetic_code = None,
endpoints = None,
entrez_query = '(none)',
expect = 10.0,
filter = None,
gapcosts = None,
genetic_code = None,
hitlist_size = 50,
i_thresh = None,
layout = None,
lcase_mask = None,
matrix_name = None,
nucl_penalty = None,
nucl_reward = None,
other_advanced = None,
perc_ident = None,
phi_pattern = None,
query_file = None,
query_believe_defline = None,
query_from = None,
query_to = None,
searchsp_eff = None,
service = None,
threshold = None,
ungapped_alignment = None,
word_size = None,
alignments = 500,
alignment_view = None,
descriptions = 500,
entrez_links_new_window = None,
expect_low = None,
expect_high = None,
format_entrez_query = None,
format_object = None,
format_type = 'XML',
ncbi_gi = None,
results_file = None,
show_overview = None,
megablast = None,
template_type = None,
template_length = None
)
BLAST search using NCBI's QBLAST server or a cloud service provider.
Supports all parameters of the qblast API for Put and Get.
Please note that BLAST on the cloud supports the NCBI-BLAST Common
URL API (http://ncbi.github.io/blast-cloud/dev/api.html).
To use this feature, please set url_base to 'http://host.my.cloud.service.provider.com/cgi-bin/blast.cgi' and
format_object = 'Alignment'. For more details, please see 8. Biopython – Overview of BLAST
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE = BlastDocs&DOC_TYPE = CloudBlast
Some useful parameters:
- program blastn, blastp, blastx, tblastn, or tblastx (lower case)
- database Which database to search against (e.g. "nr").
- sequence The sequence to search.
- ncbi_gi TRUE/FALSE whether to give 'gi' identifier.
- descriptions Number of descriptions to show. Def 500.
- alignments Number of alignments to show. Def 500.
- expect An expect value cutoff. Def 10.0.
- matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45).
- filter "none" turns off filtering. Default no filtering
- format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML".
- entrez_query Entrez query to limit Blast search
- hitlist_size Number of hits to return. Default 50
- megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only)
- service plain, psi, phi, rpsblast, megablast (lower case)
This function does no checking of the validity of the parameters
and passes the values to the server as is. More help is available at:
https://ncbi.github.io/blast-cloud/dev/api.htmlGenellikle, qblast işlevinin argümanları temelde BLAST web sayfasında ayarlayabileceğiniz farklı parametrelere benzer. Bu, qblast işlevinin anlaşılmasını kolaylaştırır ve kullanmak için öğrenme eğrisini azaltır.
Bağlanma ve Arama
BLAST çevrimiçi sürümüne bağlanma ve arama sürecini anlamak için, Biopython aracılığıyla çevrimiçi BLAST sunucusuna karşı basit bir dizi araması (yerel sıralama dosyamızda mevcuttur) yapalım.
Step 1 - adlı bir dosya oluşturun blast_example.fasta Biopython dizininde ve aşağıdaki sıra bilgisini girdi olarak verin
Example of a single sequence in FASTA/Pearson format:
>sequence A ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattcatat
tctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtc
>sequence B ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattca
tattctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtcStep 2 - NCBIWWW modülünü içe aktarın.
>>> from Bio.Blast import NCBIWWWStep 3 - Sıra dosyasını açın, blast_example.fasta python IO modülünü kullanarak.
>>> sequence_data = open("blast_example.fasta").read()
>>> sequence_data
'Example of a single sequence in FASTA/Pearson format:\n\n\n> sequence
A\nggtaagtcctctagtacaaacacccccaatattgtgatataattaaaatt
atattcatat\ntctgttgccagaaaaaacacttttaggctatattagagccatcttctttg aagcgttgtc\n\n'Step 4- Şimdi sıra verilerini ileten qblast işlevini ana parametre olarak çağırın. Diğer parametre veritabanı (nt) ve dahili programı (blastn) temsil eder.
>>> result_handle = NCBIWWW.qblast("blastn", "nt", sequence_data)
>>> result_handle
<_io.StringIO object at 0x000001EC9FAA4558>blast_resultsaramamızın sonucunu tutar. Daha sonra kullanılmak üzere bir dosyaya kaydedilebilir ve ayrıca ayrıntıları almak için ayrıştırılabilir. Bunu nasıl yapacağımızı önümüzdeki bölümde öğreneceğiz.
Step 5 - Aynı işlevsellik, aşağıda gösterildiği gibi tüm fasta dosyasını kullanmak yerine Seq nesnesi kullanılarak da yapılabilir -
>>> from Bio import SeqIO
>>> seq_record = next(SeqIO.parse(open('blast_example.fasta'),'fasta'))
>>> seq_record.id
'sequence'
>>> seq_record.seq
Seq('ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatat...gtc',
SingleLetterAlphabet())Şimdi, Seq nesnesini geçen qblast işlevini, ana parametre olarak record.seq'i çağırın.
>>> result_handle = NCBIWWW.qblast("blastn", "nt", seq_record.seq)
>>> print(result_handle)
<_io.StringIO object at 0x000001EC9FAA4558>BLAST, sekansınız için otomatik olarak bir tanımlayıcı atayacaktır.
Step 6 - result_handle nesnesi tüm sonuca sahip olacak ve daha sonra kullanılmak üzere bir dosyaya kaydedilebilir.
>>> with open('results.xml', 'w') as save_file:
>>> blast_results = result_handle.read()
>>> save_file.write(blast_results)Sonuç dosyasını nasıl ayrıştıracağımızı sonraki bölümde göreceğiz.
Bağımsız BLAST Çalıştırma
Bu bölüm BLAST'ın yerel sistemde nasıl çalıştırılacağını açıklamaktadır. BLAST'ı yerel sistemde çalıştırırsanız, daha hızlı olabilir ve ayrıca dizilere göre arama yapmak için kendi veritabanınızı oluşturmanıza olanak tanır.
BLAST bağlanıyor
Genel olarak, BLAST'ı yerel olarak çalıştırmak, büyük boyutu, yazılımı çalıştırmak için gereken ekstra çaba ve ilgili maliyet nedeniyle önerilmez. Online BLAST, temel ve ileri düzey amaçlar için yeterlidir. Elbette bazen yerel olarak yüklemeniz gerekebilir.
Çok fazla zaman ve yüksek ağ hacmi gerektirebilecek sık sık çevrimiçi aramalar yaptığınızı düşünün ve özel sıra verileriniz veya IP ile ilgili sorunlarınız varsa, yerel olarak kurmanız önerilir.
Bunu yapmak için aşağıdaki adımları izlememiz gerekiyor -
Step 1- Verilen bağlantıyı kullanarak en son patlama ikili dosyasını indirin ve kurun - ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Step 2- Aşağıdaki bağlantıyı kullanarak en son ve gerekli veritabanını indirin ve paketinden çıkarın - ftp://ftp.ncbi.nlm.nih.gov/blast/db/
BLAST yazılımı, sitelerinde çok sayıda veritabanı sağlar. Bize indirmek edelim alu.n.gz patlama veritabanı sitesinden dosyayı ve alu klasöre açmak. Bu dosya FAŞTA formatındadır. Bu dosyayı blast uygulamamızda kullanmak için önce dosyayı FAŞTA formatından blast veritabanı formatına dönüştürmemiz gerekiyor. BLAST, makeblastdb uygulamasının bu dönüşümü yapmasını sağlar.
Aşağıdaki kod parçacığını kullanın -
cd /path/to/alu
makeblastdb -in alu.n -parse_seqids -dbtype nucl -out alunYukarıdaki kodu çalıştırmak girdi dosyası alu.n'yi ayrıştıracak ve BLAST veri tabanını alun.nsq, alun.nsi vb. Çoklu dosyalar olarak oluşturacaktır. Şimdi, sırayı bulmak için bu veri tabanını sorgulayabiliriz.
BLAST'ı yerel sunucumuza kurduk ve ayrıca örnek BLAST veritabanına sahibiz, alun ona karşı sorgulamak için.
Step 3- Veritabanını sorgulamak için örnek bir sıra dosyası oluşturalım. Bir dosya search.fsa oluşturun ve aşağıdaki verileri içine koyun.
>gnl|alu|Z15030_HSAL001056 (Alu-J)
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCT
TGAGCCTAGGAGTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAA
AGAAAAAAAAAATAGCTCTGCTGGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTG
GGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCCACGATCACACCACT
GCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
>gnl|alu|D00596_HSAL003180 (Alu-Sx)
AGCCAGGTGTGGTGGCTCACGCCTGTAATCCCACCGCTTTGGGAGGCTGAGTCAGATCAC
CTGAGGTTAGGAATTTGGGACCAGCCTGGCCAACATGGCGACACCCCAGTCTCTACTAAT
AACACAAAAAATTAGCCAGGTGTGCTGGTGCATGTCTGTAATCCCAGCTACTCAGGAGGC
TGAGGCATGAGAATTGCTCACGAGGCGGAGGTTGTAGTGAGCTGAGATCGTGGCACTGTA
CTCCAGCCTGGCGACAGAGGGAGAACCCATGTCAAAAACAAAAAAAGACACCACCAAAGG
TCAAAGCATA
>gnl|alu|X55502_HSAL000745 (Alu-J)
TGCCTTCCCCATCTGTAATTCTGGCACTTGGGGAGTCCAAGGCAGGATGATCACTTATGC
CCAAGGAATTTGAGTACCAAGCCTGGGCAATATAACAAGGCCCTGTTTCTACAAAAACTT
TAAACAATTAGCCAGGTGTGGTGGTGCGTGCCTGTGTCCAGCTACTCAGGAAGCTGAGGC
AAGAGCTTGAGGCTACAGTGAGCTGTGTTCCACCATGGTGCTCCAGCCTGGGTGACAGGG
CAAGACCCTGTCAAAAGAAAGGAAGAAAGAACGGAAGGAAAGAAGGAAAGAAACAAGGAG
AGSıra verileri alu.n dosyasından toplanır; dolayısıyla, veritabanımızla eşleşir.
Step 4 - BLAST yazılımı veri tabanında arama yapmak için birçok uygulama sağlar ve biz blastn kullanırız. blastn application requires minimum of three arguments, db, query and out. db aramaya karşı veritabanını ifade eder; query eşleşecek sıradır ve outsonuçların saklanacağı dosyadır. Şimdi, bu basit sorguyu gerçekleştirmek için aşağıdaki komutu çalıştırın -
blastn -db alun -query search.fsa -out results.xml -outfmt 5Yukarıdaki komutu çalıştırmak, results.xml dosya aşağıda verildiği gibi (kısmen veri) -
<?xml version = "1.0"?>
<!DOCTYPE BlastOutput PUBLIC "-//NCBI//NCBI BlastOutput/EN"
"http://www.ncbi.nlm.nih.gov/dtd/NCBI_BlastOutput.dtd">
<BlastOutput>
<BlastOutput_program>blastn</BlastOutput_program>
<BlastOutput_version>BLASTN 2.7.1+</BlastOutput_version>
<BlastOutput_reference>Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
</BlastOutput_reference>
<BlastOutput_db>alun</BlastOutput_db>
<BlastOutput_query-ID>Query_1</BlastOutput_query-ID>
<BlastOutput_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</BlastOutput_query-def>
<BlastOutput_query-len>292</BlastOutput_query-len>
<BlastOutput_param>
<Parameters>
<Parameters_expect>10</Parameters_expect>
<Parameters_sc-match>1</Parameters_sc-match>
<Parameters_sc-mismatch>-2</Parameters_sc-mismatch>
<Parameters_gap-open>0</Parameters_gap-open>
<Parameters_gap-extend>0</Parameters_gap-extend>
<Parameters_filter>L;m;</Parameters_filter>
</Parameters>
</BlastOutput_param>
<BlastOutput_iterations>
<Iteration>
<Iteration_iter-num>1</Iteration_iter-num><Iteration_query-ID>Query_1</Iteration_query-ID>
<Iteration_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</Iteration_query-def>
<Iteration_query-len>292</Iteration_query-len>
<Iteration_hits>
<Hit>
<Hit_num>1</Hit_num>
<Hit_id>gnl|alu|Z15030_HSAL001056</Hit_id>
<Hit_def>(Alu-J)</Hit_def>
<Hit_accession>Z15030_HSAL001056</Hit_accession>
<Hit_len>292</Hit_len>
<Hit_hsps>
<Hsp>
<Hsp_num>1</Hsp_num>
<Hsp_bit-score>540.342</Hsp_bit-score>
<Hsp_score>292</Hsp_score>
<Hsp_evalue>4.55414e-156</Hsp_evalue>
<Hsp_query-from>1</Hsp_query-from>
<Hsp_query-to>292</Hsp_query-to>
<Hsp_hit-from>1</Hsp_hit-from>
<Hsp_hit-to>292</Hsp_hit-to>
<Hsp_query-frame>1</Hsp_query-frame>
<Hsp_hit-frame>1</Hsp_hit-frame>
<Hsp_identity>292</Hsp_identity>
<Hsp_positive>292</Hsp_positive>
<Hsp_gaps>0</Hsp_gaps>
<Hsp_align-len>292</Hsp_align-len>
<Hsp_qseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGAGTTTG
CGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCTGGTGGTGCATG
CCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCC
ACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
</Hsp_qseq>
<Hsp_hseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGA
GTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCT
GGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGG
CTGTGGTGAGCCACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAAC
AAATAA
</Hsp_hseq>
<Hsp_midline>
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||
</Hsp_midline>
</Hsp>
</Hit_hsps>
</Hit>
.........................
.........................
.........................
</Iteration_hits>
<Iteration_stat>
<Statistics>
<Statistics_db-num>327</Statistics_db-num>
<Statistics_db-len>80506</Statistics_db-len>
<Statistics_hsp-lenv16</Statistics_hsp-len>
<Statistics_eff-space>21528364</Statistics_eff-space>
<Statistics_kappa>0.46</Statistics_kappa>
<Statistics_lambda>1.28</Statistics_lambda>
<Statistics_entropy>0.85</Statistics_entropy>
</Statistics>
</Iteration_stat>
</Iteration>
</BlastOutput_iterations>
</BlastOutput>Yukarıdaki komut python içinde aşağıdaki kod kullanılarak çalıştırılabilir -
>>> from Bio.Blast.Applications import NcbiblastnCommandline
>>> blastn_cline = NcbiblastnCommandline(query = "search.fasta", db = "alun",
outfmt = 5, out = "results.xml")
>>> stdout, stderr = blastn_cline()Burada birincisi, püskürtme çıktısının bir tutamağıdır ve ikincisi, püskürtme komutu tarafından üretilen olası hata çıktısıdır.
Çıktı dosyasını komut satırı bağımsız değişkeni (out = "results.xml") olarak sağladığımız ve çıktı biçimini XML olarak ayarladığımızdan (outfmt = 5), çıktı dosyası geçerli çalışma dizinine kaydedilecektir.
BLAST Sonucunun Ayrıştırılması
Genellikle, BLAST çıktısı NCBIXML modülü kullanılarak XML formatı olarak ayrıştırılır. Bunu yapmak için aşağıdaki modülü içe aktarmamız gerekiyor -
>>> from Bio.Blast import NCBIXMLŞimdi, open the file directly using python open method ve use NCBIXML parse method aşağıda verildiği gibi -
>>> E_VALUE_THRESH = 1e-20
>>> for record in NCBIXML.parse(open("results.xml")):
>>> if record.alignments:
>>> print("\n")
>>> print("query: %s" % record.query[:100])
>>> for align in record.alignments:
>>> for hsp in align.hsps:
>>> if hsp.expect < E_VALUE_THRESH:
>>> print("match: %s " % align.title[:100])Bu, aşağıdaki gibi bir çıktı üretecektir -
query: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|L12964_HSAL003860 (Alu-J)
match: gnl|alu|L13042_HSAL003863 (Alu-FLA?)
match: gnl|alu|M86249_HSAL001462 (Alu-FLA?)
match: gnl|alu|M29484_HSAL002265 (Alu-J)
query: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|J03071_HSAL001860 (Alu-J)
match: gnl|alu|X72409_HSAL005025 (Alu-Sx)
query: gnl|alu|X55502_HSAL000745 (Alu-J)
match: gnl|alu|X55502_HSAL000745 (Alu-J)EntrezNCBI tarafından sağlanan bir çevrimiçi arama sistemidir. Boole operatörlerini ve alan aramasını destekleyen entegre bir küresel sorgu ile neredeyse tüm bilinen moleküler biyoloji veritabanlarına erişim sağlar. Her veritabanından gelen isabet sayısı, kaynak veritabanına bağlantılar içeren kayıtlar, vb. Gibi bilgilerle tüm veritabanlarından sonuçları döndürür.
Entrez aracılığıyla erişilebilen popüler veri tabanlarından bazıları aşağıda listelenmiştir -
- Pubmed
- Pubmed Central
- Nükleotid (GenBank Dizi Veritabanı)
- Protein (Dizi Veritabanı)
- Genom (Tüm Genom Veritabanı)
- Yapı (Üç Boyutlu Makromoleküler Yapı)
- Taksonomi (GenBank'taki Organizmalar)
- SNP (Tek Nükleotid Polimorfizmi)
- UniGene (Gene Yönelik Transkript Dizileri Kümeleri)
- CDD (Korunmuş Protein Alan Veritabanı)
- 3B Etki Alanları (Entrez Yapısından Alanlar)
Entrez, yukarıdaki veri tabanlarına ek olarak, alan araştırması gerçekleştirmek için daha birçok veri tabanı sağlar.
Biopython, Entrez veritabanına erişmek için Entrez'e özel bir modül olan Bio.Entrez sağlar. Bu bölümde Biopython kullanarak Entrez'e nasıl erişeceğimizi öğrenelim -
Veritabanı Bağlantı Adımları
Entrez'in özelliklerini eklemek için aşağıdaki modülü içe aktarın -
>>> from Bio import EntrezArdından, aşağıda verilen koda kimin bağlı olduğunu belirlemek için e-postanızı ayarlayın -
>>> Entrez.email = '<youremail>'Ardından, Entrez aracı parametresini ayarlayın ve varsayılan olarak Biopython'dur.
>>> Entrez.tool = 'Demoscript'Şimdi, call einfo function to find index term counts, last update, and available links for each database aşağıda tanımlandığı gibi -
>>> info = Entrez.einfo()Einfo yöntemi, aşağıda gösterildiği gibi okuma yöntemi aracılığıyla bilgilere erişim sağlayan bir nesne döndürür -
>>> data = info.read()
>>> print(data)
<?xml version = "1.0" encoding = "UTF-8" ?>
<!DOCTYPE eInfoResult PUBLIC "-//NLM//DTD einfo 20130322//EN"
"https://eutils.ncbi.nlm.nih.gov/eutils/dtd/20130322/einfo.dtd">
<eInfoResult>
<DbList>
<DbName>pubmed</DbName>
<DbName>protein</DbName>
<DbName>nuccore</DbName>
<DbName>ipg</DbName>
<DbName>nucleotide</DbName>
<DbName>nucgss</DbName>
<DbName>nucest</DbName>
<DbName>structure</DbName>
<DbName>sparcle</DbName>
<DbName>genome</DbName>
<DbName>annotinfo</DbName>
<DbName>assembly</DbName>
<DbName>bioproject</DbName>
<DbName>biosample</DbName>
<DbName>blastdbinfo</DbName>
<DbName>books</DbName>
<DbName>cdd</DbName>
<DbName>clinvar</DbName>
<DbName>clone</DbName>
<DbName>gap</DbName>
<DbName>gapplus</DbName>
<DbName>grasp</DbName>
<DbName>dbvar</DbName>
<DbName>gene</DbName>
<DbName>gds</DbName>
<DbName>geoprofiles</DbName>
<DbName>homologene</DbName>
<DbName>medgen</DbName>
<DbName>mesh</DbName>
<DbName>ncbisearch</DbName>
<DbName>nlmcatalog</DbName>
<DbName>omim</DbName>
<DbName>orgtrack</DbName>
<DbName>pmc</DbName>
<DbName>popset</DbName>
<DbName>probe</DbName>
<DbName>proteinclusters</DbName>
<DbName>pcassay</DbName>
<DbName>biosystems</DbName>
<DbName>pccompound</DbName>
<DbName>pcsubstance</DbName>
<DbName>pubmedhealth</DbName>
<DbName>seqannot</DbName>
<DbName>snp</DbName>
<DbName>sra</DbName>
<DbName>taxonomy</DbName>
<DbName>biocollections</DbName>
<DbName>unigene</DbName>
<DbName>gencoll</DbName>
<DbName>gtr</DbName>
</DbList>
</eInfoResult>Veriler XML biçimindedir ve verileri python nesnesi olarak almak için şunu kullanın: Entrez.read en kısa sürede yöntem Entrez.einfo() yöntem çağrılır -
>>> info = Entrez.einfo()
>>> record = Entrez.read(info)Burada, kayıt, aşağıda gösterildiği gibi bir anahtarı olan DbList olan bir sözlüktür -
>>> record.keys()
[u'DbList']DbList anahtarına erişim, aşağıda gösterilen veritabanı adlarının listesini döndürür -
>>> record[u'DbList']
['pubmed', 'protein', 'nuccore', 'ipg', 'nucleotide', 'nucgss',
'nucest', 'structure', 'sparcle', 'genome', 'annotinfo', 'assembly',
'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar',
'clone', 'gap', 'gapplus', 'grasp', 'dbvar', 'gene', 'gds', 'geoprofiles',
'homologene', 'medgen', 'mesh', 'ncbisearch', 'nlmcatalog', 'omim',
'orgtrack', 'pmc', 'popset', 'probe', 'proteinclusters', 'pcassay',
'biosystems', 'pccompound', 'pcsubstance', 'pubmedhealth', 'seqannot',
'snp', 'sra', 'taxonomy', 'biocollections', 'unigene', 'gencoll', 'gtr']
>>>Temel olarak Entrez modülü, Entrez arama sistemi tarafından döndürülen XML'i ayrıştırır ve python sözlüğü ve listeleri olarak sağlar.
Veritabanında Ara
Entrez veritabanlarından herhangi birini aramak için Bio.Entrez.esearch () modülünü kullanabiliriz. Aşağıda tanımlanmıştır -
>>> info = Entrez.einfo()
>>> info = Entrez.esearch(db = "pubmed",term = "genome")
>>> record = Entrez.read(info)
>>>print(record)
DictElement({u'Count': '1146113', u'RetMax': '20', u'IdList':
['30347444', '30347404', '30347317', '30347292',
'30347286', '30347249', '30347194', '30347187',
'30347172', '30347088', '30347075', '30346992',
'30346990', '30346982', '30346980', '30346969',
'30346962', '30346954', '30346941', '30346939'],
u'TranslationStack': [DictElement({u'Count':
'927819', u'Field': 'MeSH Terms', u'Term': '"genome"[MeSH Terms]',
u'Explode': 'Y'}, attributes = {})
, DictElement({u'Count': '422712', u'Field':
'All Fields', u'Term': '"genome"[All Fields]', u'Explode': 'N'}, attributes = {}),
'OR', 'GROUP'], u'TranslationSet': [DictElement({u'To': '"genome"[MeSH Terms]
OR "genome"[All Fields]', u'From': 'genome'}, attributes = {})], u'RetStart': '0',
u'QueryTranslation': '"genome"[MeSH Terms] OR "genome"[All Fields]'},
attributes = {})
>>>Yanlış db atarsanız, geri döner
>>> info = Entrez.esearch(db = "blastdbinfo",term = "books")
>>> record = Entrez.read(info)
>>> print(record)
DictElement({u'Count': '0', u'RetMax': '0', u'IdList': [],
u'WarningList': DictElement({u'OutputMessage': ['No items found.'],
u'PhraseIgnored': [], u'QuotedPhraseNotFound': []}, attributes = {}),
u'ErrorList': DictElement({u'FieldNotFound': [], u'PhraseNotFound':
['books']}, attributes = {}), u'TranslationSet': [], u'RetStart': '0',
u'QueryTranslation': '(books[All Fields])'}, attributes = {})Veritabanında arama yapmak istiyorsanız, Entrez.egquery. Bu benzerEntrez.esearch ancak anahtar kelimeyi belirtmek ve veritabanı parametresini atlamak yeterlidir.
>>>info = Entrez.egquery(term = "entrez")
>>> record = Entrez.read(info)
>>> for row in record["eGQueryResult"]:
... print(row["DbName"], row["Count"])
...
pubmed 458
pmc 12779 mesh 1
...
...
...
biosample 7
biocollections 0Kayıtları Al
Enterz, Entrez'den bir kaydın tüm ayrıntılarını aramak ve indirmek için özel bir yöntem, efetch sağlar. Aşağıdaki basit örneği düşünün -
>>> handle = Entrez.efetch(
db = "nucleotide", id = "EU490707", rettype = "fasta")Şimdi, SeqIO nesnesini kullanarak kayıtları kolayca okuyabiliriz
>>> record = SeqIO.read( handle, "fasta" )
>>> record
SeqRecord(seq = Seq('ATTTTTTACGAACCTGTGGAAATTTTTGGTTATGACAATAAATCTAGTTTAGTA...GAA',
SingleLetterAlphabet()), id = 'EU490707.1', name = 'EU490707.1',
description = 'EU490707.1
Selenipedium aequinoctiale maturase K (matK) gene, partial cds; chloroplast',
dbxrefs = [])Biopython, polipeptit yapılarını manipüle etmek için Bio.PDB modülü sağlar. PDB (Protein Veri Bankası), çevrimiçi olarak mevcut en büyük protein yapısı kaynağıdır. Protein-protein, protein-DNA, protein-RNA kompleksleri dahil birçok farklı protein yapısına ev sahipliği yapar.
PDB'yi yüklemek için aşağıdaki komutu yazın -
from Bio.PDB import *Protein Yapısı Dosya Biçimleri
PDB, protein yapılarını üç farklı formatta dağıtır -
- Biopython tarafından desteklenmeyen XML tabanlı dosya biçimi
- Özel olarak biçimlendirilmiş bir metin dosyası olan pdb dosya biçimi
- PDBx / mmCIF dosya biçimi
Protein Veri Bankası tarafından dağıtılan PDB dosyaları, belirsiz veya ayrıştırılmalarını zorlaştıran biçimlendirme hataları içerebilir. Bio.PDB modülü bu hatalarla otomatik olarak ilgilenmeye çalışır.
Bio.PDB modülü, biri mmCIF formatı ve ikincisi pdb formatı olmak üzere iki farklı ayrıştırıcı uygular.
Biçimlerin her birini ayrıntılı olarak nasıl ayrıştıracağımızı öğrenelim -
mmCIF Ayrıştırıcı
Aşağıdaki komutu kullanarak pdb sunucusundan mmCIF formatında örnek bir veritabanı indirelim -
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'mmCif')Bu, belirtilen dosyayı (2fat.cif) sunucudan indirecek ve mevcut çalışma dizininde saklayacaktır.
Burada, PDBList, dosyaları çevrimiçi PDB FTP sunucusundan listelemek ve indirmek için seçenekler sunar. retrieve_pdb_file yöntemi, uzantısız indirilecek dosyanın adına ihtiyaç duyar. retrieve_pdb_file ayrıca indirme dizinini, pdir ve dosyanın biçimini, file_format'ı belirtme seçeneğine de sahiptir. Dosya formatının olası değerleri aşağıdaki gibidir -
- "MmCif" (varsayılan, PDBx / mmCif dosyası)
- "Pdb" (PDB biçimi)
- "Xml" (PMDML / XML biçimi)
- "Mmtf" (yüksek oranda sıkıştırılmış)
- "Paket" (büyük yapı için PDB formatlı arşiv)
Bir cif dosyası yüklemek için Bio.MMCIF.MMCIFParser'ı aşağıda belirtildiği gibi kullanın -
>>> parser = MMCIFParser(QUIET = True)
>>> data = parser.get_structure("2FAT", "2FAT.cif")Burada QUIET, dosyayı ayrıştırırken uyarıyı bastırır. get_structure will parse the file and return the structure with id as 2FAT (ilk argüman).
Yukarıdaki komutu çalıştırdıktan sonra, dosyayı ayrıştırır ve varsa olası uyarıyı yazdırır.
Şimdi, aşağıdaki komutu kullanarak yapıyı kontrol edin -
>>> data
<Structure id = 2FAT>
To get the type, use type method as specified below,
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Dosyayı başarıyla ayrıştırdık ve proteinin yapısını aldık. Protein yapısının ayrıntılarını ve onu nasıl elde edeceğimizi sonraki bölümde öğreneceğiz.
PDB Ayrıştırıcı
Aşağıdaki komutu kullanarak pdb sunucusundan PDB formatında örnek bir veritabanı indirelim -
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'pdb')Bu, belirtilen dosyayı (pdb2fat.ent) sunucudan indirecek ve mevcut çalışma dizininde saklayacaktır.
Bir pdb dosyası yüklemek için Bio.PDB.PDBParser'ı aşağıda belirtildiği gibi kullanın -
>>> parser = PDBParser(PERMISSIVE = True, QUIET = True)
>>> data = parser.get_structure("2fat","pdb2fat.ent")Burada get_structure, MMCIFParser'a benzer. PERMISSIVE seçeneği, protein verilerini mümkün olduğunca esnek şekilde ayrıştırmaya çalışın.
Şimdi, yapıyı ve türünü aşağıda verilen kod parçacığı ile kontrol edin -
>>> data
<Structure id = 2fat>
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Başlık yapısı sözlük bilgilerini saklar. Bunu gerçekleştirmek için aşağıdaki komutu yazın -
>>> print(data.header.keys()) dict_keys([
'name', 'head', 'deposition_date', 'release_date', 'structure_method', 'resolution',
'structure_reference', 'journal_reference', 'author', 'compound', 'source',
'keywords', 'journal'])
>>>İsmi almak için aşağıdaki kodu kullanın -
>>> print(data.header["name"])
an anti-urokinase plasminogen activator receptor (upar) antibody: crystal
structure and binding epitope
>>>Aşağıdaki kodla tarihi ve çözünürlüğü de kontrol edebilirsiniz -
>>> print(data.header["release_date"]) 2006-11-14
>>> print(data.header["resolution"]) 1.77PDB Yapısı
PDB yapısı, iki zincir içeren tek bir modelden oluşur.
- kalıntı sayısını içeren zincir L
- kalıntı sayısı içeren zincir H
Her kalıntı, her biri (x, y, z) koordinatlarıyla temsil edilen bir 3D konuma sahip olan birden çok atomdan oluşur.
Atomun yapısını nasıl elde edeceğimizi aşağıdaki bölümde detaylı olarak öğrenelim -
Modeli
Structure.get_models () yöntemi, modeller üzerinde bir yineleyici döndürür. Aşağıda tanımlanmıştır -
>>> model = data.get_models()
>>> model
<generator object get_models at 0x103fa1c80>
>>> models = list(model)
>>> models [<Model id = 0>]
>>> type(models[0])
<class 'Bio.PDB.Model.Model'>Burada, bir Model tam olarak bir 3B konformasyonu tanımlar. Bir veya daha fazla zincir içerir.
Zincir
Model.get_chain () yöntemi, zincirler üzerinde bir yineleyici döndürür. Aşağıda tanımlanmıştır -
>>> chains = list(models[0].get_chains())
>>> chains
[<Chain id = L>, <Chain id = H>]
>>> type(chains[0])
<class 'Bio.PDB.Chain.Chain'>Burada Zincir, uygun bir polipeptit yapısını, yani ardışık bir bağlı kalıntı dizisini tarif eder.
Kalıntı
Chain.get_residues () yöntemi, kalıntılar üzerinde bir yineleyici döndürür. Aşağıda tanımlanmıştır -
>>> residue = list(chains[0].get_residues())
>>> len(residue)
293
>>> residue1 = list(chains[1].get_residues())
>>> len(residue1)
311Residue, bir amino aside ait atomları tutar.
Atomlar
Residue.get_atom (), aşağıda tanımlandığı gibi atomlar üzerinde bir yineleyici döndürür -
>>> atoms = list(residue[0].get_atoms())
>>> atoms
[<Atom N>, <Atom CA>, <Atom C>, <Atom Ov, <Atom CB>, <Atom CG>, <Atom OD1>, <Atom OD2>]Bir atom, bir atomun 3B koordinatını tutar ve buna Vektör adı verilir. Aşağıda tanımlanmıştır
>>> atoms[0].get_vector()
<Vector 18.49, 73.26, 44.16>X, y ve z koordinat değerlerini temsil eder.
Bir dizi motifi, bir nükleotid veya amino asit dizisi modelidir. Sekans motifleri, bitişik olmayabilen amino asitlerin üç boyutlu düzenlenmesiyle oluşturulur. Biopython, aşağıda belirtilen sekans motifinin işlevlerine erişmek için ayrı bir modül olan Bio.motifs sağlar -
from Bio import motifsBasit DNA Motifi Oluşturmak
Aşağıdaki komutu kullanarak basit bir DNA motif dizisi oluşturalım -
>>> from Bio import motifs
>>> from Bio.Seq import Seq
>>> DNA_motif = [ Seq("AGCT"),
... Seq("TCGA"),
... Seq("AACT"),
... ]
>>> seq = motifs.create(DNA_motif)
>>> print(seq) AGCT TCGA AACTSıra değerlerini saymak için aşağıdaki komutu kullanın -
>>> print(seq.counts)
0 1 2 3
A: 2.00 1.00 0.00 1.00
C: 0.00 1.00 2.00 0.00
G: 0.00 1.00 1.00 0.00
T: 1.00 0.00 0.00 2.00Sıradaki 'A'yı saymak için aşağıdaki kodu kullanın -
>>> seq.counts["A", :]
(2, 1, 0, 1)Sayım sütunlarına erişmek istiyorsanız, aşağıdaki komutu kullanın -
>>> seq.counts[:, 3]
{'A': 1, 'C': 0, 'T': 2, 'G': 0}Sıralı Logo Oluşturma
Şimdi bir Sıra Logosunun nasıl oluşturulacağını tartışacağız.
Aşağıdaki sırayı düşünün -
AGCTTACG
ATCGTACC
TTCCGAAT
GGTACGTA
AAGCTTGGAşağıdaki bağlantıyı kullanarak kendi logonuzu oluşturabilirsiniz - http://weblogo.berkeley.edu/
Yukarıdaki diziyi ekleyin ve yeni bir logo oluşturun ve seq.png adlı resmi biopython klasörünüze kaydedin.
seq.pngGörüntüyü oluşturduktan sonra, şimdi aşağıdaki komutu çalıştırın -
>>> seq.weblogo("seq.png")Bu DNA dizisi motifi, LexA bağlama motifi için bir sekans logosu olarak temsil edilir.
JASPAR Veritabanı
JASPAR, en popüler veri tabanlarından biridir. Dizileri okumak, yazmak ve taramak için herhangi bir motif formatının kolaylığını sağlar. Her motif için meta bilgileri depolar.The module Bio.motifs contains a specialized class jaspar.Motif to represent meta-information attributes.
Aşağıdaki dikkate değer nitelik türlerine sahiptir -
- matrix_id - Benzersiz JASPAR motif kimliği
- isim - Motifin adı
- tf_family - Motif ailesi, örneğin 'Helix-Loop-Helix'
- veri_türü - motifte kullanılan veri türü.
Biopython klasöründe sample.sites içinde adlandırılan bir JASPAR site formatı oluşturalım. Aşağıda tanımlanmıştır -
sample.sites
>MA0001 ARNT 1
AACGTGatgtccta
>MA0001 ARNT 2
CAGGTGggatgtac
>MA0001 ARNT 3
TACGTAgctcatgc
>MA0001 ARNT 4
AACGTGacagcgct
>MA0001 ARNT 5
CACGTGcacgtcgt
>MA0001 ARNT 6
cggcctCGCGTGcYukarıdaki dosyada motif örnekleri oluşturduk. Şimdi, yukarıdaki örneklerden bir motif nesnesi oluşturalım -
>>> from Bio import motifs
>>> with open("sample.sites") as handle:
... data = motifs.read(handle,"sites")
...
>>> print(data)
TF name None
Matrix ID None
Matrix:
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00Burada veriler, sample.sites dosyasındaki tüm motif örneklerini okur.
Verilerden tüm örnekleri yazdırmak için aşağıdaki komutu kullanın -
>>> for instance in data.instances:
... print(instance)
...
AACGTG
CAGGTG
TACGTA
AACGTG
CACGTG
CGCGTGTüm değerleri saymak için aşağıdaki komutu kullanın -
>>> print(data.counts)
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00
>>>BioSQLesas olarak tüm RDBMS motoru için dizileri ve ilgili verilerini depolamak için tasarlanmış genel bir veritabanı şemasıdır. GenBank, Swissport, vb. Gibi tüm popüler biyoinformatik veri tabanlarından verileri tutacak şekilde tasarlanmıştır. Şirket içi verileri depolamak için de kullanılabilir.
BioSQL şu anda aşağıdaki veritabanları için özel şema sağlamaktadır -
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Ayrıca Java tabanlı HSQLDB ve Derby veritabanları için minimum destek sağlar.
BioPython, BioSQL tabanlı veritabanıyla çalışmak için çok basit, kolay ve gelişmiş ORM yetenekleri sağlar. BioPython provides a module, BioSQL aşağıdaki işlevi yapmak için -
- BioSQL veritabanı oluştur / kaldır
- BioSQL veritabanına bağlanın
- GenBank, Swisport, BLAST sonucu, Entrez sonucu gibi bir dizi veritabanını ayrıştırın ve doğrudan BioSQL veritabanına yükleyin
- BioSQL veritabanından sekans verilerini alın
- Sınıflandırma verilerini NCBI BLAST'tan alın ve BioSQL veritabanında saklayın
- BioSQL veritabanında herhangi bir SQL sorgusu çalıştırın
BioSQL Veritabanı Şemasına Genel Bakış
BioSQL'in derinliklerine inmeden önce, BioSQL şemasının temellerini anlayalım. BioSQL şeması, sekans verilerini, sekans özelliğini, sekans kategorisi / ontolojisini ve taksonomi bilgilerini tutmak için 25'ten fazla tablo sağlar. Önemli tablolardan bazıları aşağıdaki gibidir -
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
BioSQL Veritabanı Oluşturma
Bu bölümde, BioSQL ekibi tarafından sağlanan şemayı kullanarak örnek bir BioSQL veritabanı, biosql oluşturalım. Başlamak gerçekten çok kolay olduğundan ve karmaşık bir kurulum içermediğinden SQLite veritabanı ile çalışacağız.
Burada, aşağıdaki adımları kullanarak SQLite tabanlı bir BioSQL veritabanı oluşturacağız.
Step 1 - SQLite veritabanı motorunu indirin ve kurun.
Step 2 - BioSQL projesini GitHub URL'sinden indirin. https://github.com/biosql/biosql
Step 3 - Bir konsol açın ve mkdir kullanarak bir dizin oluşturun ve içine girin.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 - Yeni bir SQLite veritabanı oluşturmak için aşağıdaki komutu çalıştırın.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 - Biosqldb-sqlite.sql dosyasını BioSQL projesinden (/ sql / biosqldb-sqlite.sql`) kopyalayın ve mevcut dizinde saklayın.
Step 6 - Tüm tabloları oluşturmak için aşağıdaki komutu çalıştırın.
sqlite> .read biosqldb-sqlite.sqlArtık tüm tablolar yeni veritabanımızda oluşturulmuştur.
Step 7 - Veritabanımızdaki tüm yeni tabloları görmek için aşağıdaki komutu çalıştırın.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>İlk üç komut, sonucu biçimlendirilmiş bir şekilde göstermek için SQLite'ı yapılandırmak için yapılandırma komutlarıdır.
Step 8 - BioPython ekibi tarafından sağlanan ls_orchid.gbk örnek GenBank dosyasını kopyalayın https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk geçerli dizine ve orchid.gbk olarak kaydedin.
Step 9 - Aşağıdaki kodu kullanarak bir python betiği, load_orchid.py oluşturun ve çalıştırın.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Yukarıdaki kod, dosyadaki kaydı ayrıştırır ve onu python nesnelerine dönüştürür ve BioSQL veritabanına ekler. Kodu sonraki bölümde analiz edeceğiz.
Son olarak, yeni bir BioSQL veritabanı oluşturduk ve içine bazı örnek veriler yükledik. Önümüzdeki bölümde önemli tabloları tartışacağız.
Basit ER Diyagramı
biodatabase tablo, hiyerarşinin en üstündedir ve ana amacı, bir dizi veri dizisini tek bir grup / sanal veritabanında organize etmektir. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. BioSQL veritabanındaki tüm ilgili tabloların biyo veritabanı girişine referansları vardır.
bioentrytablo, sıra verileri dışında bir diziyle ilgili tüm ayrıntıları içerir. belirli bir dizilim verileribioentry depolanacak biosequence tablo.
takson ve takson_adı, taksonomi ayrıntılarıdır ve her giriş, takson bilgilerini belirtmek için bu tabloya başvurur.
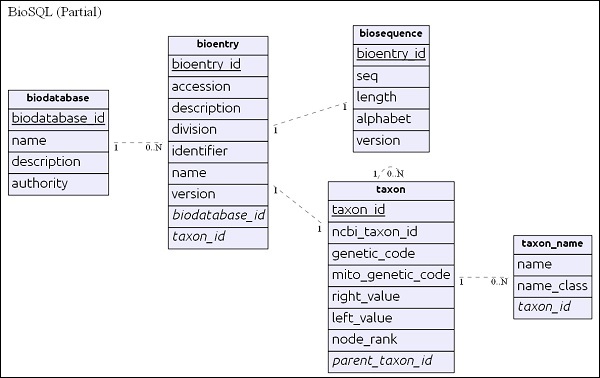
Şemayı anladıktan sonra, sonraki bölümde bazı sorgulara bakalım.
BioSQL Sorguları
Verilerin nasıl düzenlendiğini ve tabloların birbiriyle nasıl ilişkili olduğunu daha iyi anlamak için bazı SQL sorgularını inceleyelim. Devam etmeden önce, aşağıdaki komutu kullanarak veritabanını açalım ve bazı biçimlendirme komutlarını ayarlayalım -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Sorguyu çalıştırmak için herhangi bir SQLite düzenleyicisini de kullanabilirsiniz.
Sistemde bulunan sanal sıra veritabanını aşağıda verildiği gibi listeleyin -
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Burada sadece bir veritabanımız var, orchid.
Veritabanında bulunan girişleri (ilk 3) listeleyin orchid aşağıda verilen kod ile
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Bir girişle ilişkili sekans ayrıntılarını (erişim - Z78530, isim - C. fasciculatum 5.8S rRNA geni ve ITS1 ve ITS2 DNA) verilen kodla listeleyin -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Aşağıdaki kodu kullanarak bir girişle ilişkili tam diziyi alın (erişim - Z78530, isim - C. fasciculatum 5.8S rRNA geni ve ITS1 ve ITS2 DNA) -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Bio veritabanı, orkide ile ilişkili taksonları listeleyin
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>BioSQL Veritabanına Veri Yükleme
Bu bölümde BioSQL veritabanına sekans verilerini nasıl yükleyeceğimizi öğrenelim. Önceki bölümde veri tabanına veri yüklemek için koda zaten sahibiz ve kod aşağıdaki gibidir -
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Kodun her satırına ve amacına daha derinlemesine bakacağız -
Line 1 - SeqIO modülünü yükler.
Line 2- BioSeqDatabase modülünü yükler. Bu modül, BioSQL veritabanı ile etkileşim için tüm işlevselliği sağlar.
Line 3 - OS modülünü yükler.
Line 5- open_database, yapılandırılmış sürücü (sürücü) ile belirtilen veritabanını (db) açar ve BioSQL veritabanına (sunucu) bir tutamaç döndürür. Biopython sqlite, mysql, postgresql ve oracle veritabanlarını destekler.
Line 6-10- load_database_sql yöntemi, sql'yi harici dosyadan yükler ve çalıştırır. commit yöntemi işlemi taahhüt eder. Veritabanını zaten şemayla oluşturduğumuz için bu adımı atlayabiliriz.
Line 12 - new_database yöntemleri, yeni sanal veritabanı, orkide oluşturur ve Orchid veritabanına karşı komutu yürütmek için bir tutamaç veritabanı döndürür.
Line 13- yükleme yöntemi, sıralama girdilerini (yinelenebilir Sıralı Kayıt) orkide veritabanına yükler. SqlIO.parse, GenBank veritabanını ayrıştırır ve içindeki tüm dizileri yinelenebilir SeqRecord olarak döndürür. Yükleme yönteminin ikinci parametresi (True), sistemde halihazırda mevcut değilse, NCBI blast web sitesinden dizi verilerinin taksonomi ayrıntılarını alması talimatını verir.
Line 14 - commit işlemi taahhüt eder.
Line 15 - kapat, veritabanı bağlantısını kapatır ve sunucu tutamacını yok eder.
Sıra Verilerini Getir
Orchid veritabanından tanımlayıcı 2765658 olan bir sekansı aşağıdaki gibi getirelim -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Burada, sunucu ["orkide"], sanal veritabanından veyachid'den veri almak için tanıtıcıyı döndürür. lookup yöntem, kriterlere göre dizileri seçme seçeneği sunar ve 2765658 tanımlayıcılı diziyi seçtik. lookupsekans bilgisini SeqRecordobject olarak döndürür. SeqRecord` ile nasıl çalışılacağını zaten bildiğimiz için, ondan veri almak çok kolay.
Veritabanını Kaldır
Bir veritabanını kaldırmak, uygun veritabanı adıyla remove_database yöntemini çağırmak ve ardından bunu aşağıda belirtildiği gibi uygulamak kadar basittir -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()Popülasyon genetiği, evrim teorisinde önemli bir rol oynar. Türler arasındaki genetik farkı ve aynı türdeki iki veya daha fazla bireyi analiz eder.
Biopython, popülasyon genetiği için Bio.PopGen modülü sağlar ve esas olarak Michel Raymond ve Francois Rousset tarafından geliştirilen popüler bir genetik paketi olan `GenePop'u destekler.
Basit bir ayrıştırıcı
GenePop formatını ayrıştırmak ve kavramı anlamak için basit bir uygulama yazalım.
Aşağıda verilen bağlantıdan Biopython ekibi tarafından sağlanan genePop dosyasını indirin -https://raw.githubusercontent.com/biopython/biopython/master/Tests/PopGen/c3line.gen
Aşağıdaki kod parçacığını kullanarak GenePop modülünü yükleyin -
from Bio.PopGen import GenePopAşağıdaki gibi GenePop.read yöntemini kullanarak dosyayı ayrıştırın -
record = GenePop.read(open("c3line.gen"))Lokus ve popülasyon bilgilerini aşağıda verildiği gibi gösterin -
>>> record.loci_list
['136255903', '136257048', '136257636']
>>> record.pop_list
['4', 'b3', '5']
>>> record.populations
[[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (3, 4), (2, 2)]),
('3', [(3, 3), (4, 4), (2, 2)]), ('4', [(3, 3), (4, 3), (None, None)])],
[('b1', [(None, None), (4, 4), (2, 2)]), ('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]), ('4',
[(None, None), (4, 4), (2, 2)]), ('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Burada, dosyada mevcut üç lokus ve üç set popülasyon vardır: İlk popülasyonda 4 kayıt, ikinci popülasyonda 3 kayıt ve üçüncü popülasyonda 5 kayıt vardır. kayıt. popülasyonlar, her lokus için alel verileriyle tüm popülasyon kümelerini gösterir.
GenePop dosyasını işleyin
Biopython, lokus ve popülasyon verilerini kaldırmak için seçenekler sunar.
Remove a population set by position,
>>> record.remove_population(0)
>>> record.populations
[[('b1', [(None, None), (4, 4), (2, 2)]),
('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]),
('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]),
('4', [(None, None), (4, 4), (2, 2)]),
('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Remove a locus by position,
>>> record.remove_locus_by_position(0)
>>> record.loci_list
['136257048', '136257636']
>>> record.populations
[[('b1', [(4, 4), (2, 2)]), ('b2', [(4, 4), (2, 2)]), ('b3', [(4, 4), (2, 2)])],
[('1', [(4, 4), (2, 2)]), ('2', [(1, 4), (2, 2)]),
('3', [(1, 1), (2, 2)]), ('4', [(4, 4), (2, 2)]), ('5', [(4, 4), (2, 2)])]]
>>>Remove a locus by name,
>>> record.remove_locus_by_name('136257636') >>> record.loci_list
['136257048']
>>> record.populations
[[('b1', [(4, 4)]), ('b2', [(4, 4)]), ('b3', [(4, 4)])],
[('1', [(4, 4)]), ('2', [(1, 4)]),
('3', [(1, 1)]), ('4', [(4, 4)]), ('5', [(4, 4)])]]
>>>GenePop Yazılımı ile Arayüz
Biopython, GenePop yazılımıyla etkileşime geçmek için arayüzler sağlar ve böylece ondan birçok işlevselliği ortaya çıkarır. Bio.PopGen.GenePop modülü bu amaçla kullanılmaktadır. Kolay kullanımlı bir arayüz EasyController'dır. GenePop dosyasını nasıl ayrıştıracağımızı kontrol edelim ve EasyController'ı kullanarak bazı analizler yapalım.
İlk olarak, GenePop yazılımını kurun ve kurulum klasörünü sistem yoluna yerleştirin. GenePop dosyası hakkında temel bilgileri almak için, bir EasyController nesnesi oluşturun ve ardından aşağıda belirtildiği gibi get_basic_info yöntemini çağırın -
>>> from Bio.PopGen.GenePop.EasyController import EasyController
>>> ec = EasyController('c3line.gen')
>>> print(ec.get_basic_info())
(['4', 'b3', '5'], ['136255903', '136257048', '136257636'])
>>>Burada ilk öğe popülasyon listesi ve ikinci öğe lokus listesidir.
Belirli bir lokusun tüm alel listesini almak için, lokus adını aşağıda belirtildiği gibi ileterek get_alleles_all_pops yöntemini çağırın -
>>> allele_list = ec.get_alleles_all_pops("136255903")
>>> print(allele_list)
[2, 3]Alel listesini belirli bir popülasyon ve lokusa göre almak için, aşağıda verildiği gibi lokus adını ve popülasyon pozisyonunu ileterek get_alleles çağırın -
>>> allele_list = ec.get_alleles(0, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(1, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(2, "136255903")
>>> print(allele_list)
[2, 3]
>>>Benzer şekilde, EasyController birçok işlevi ortaya çıkarır: alel frekansı, genotip frekansı, çok odaklı F istatistikleri, Hardy-Weinberg dengesi, Bağlantı Dengesizliği, vb.
Bir genom, tüm genleri de dahil olmak üzere eksiksiz bir DNA kümesidir. Genom analizi, bireysel genlerin ve bunların kalıtımdaki rollerinin incelenmesini ifade eder.
Genom Diyagramı
Genom diyagramı, genetik bilgiyi grafikler halinde temsil eder. Biopython, GenomeDiagram'ı temsil etmek için Bio.Graphics.GenomeDiagram modülünü kullanır. GenomeDiagram modülü, ReportLab'in kurulu olmasını gerektirir.
Diyagram oluşturma adımları
Bir diyagram oluşturma süreci genellikle aşağıdaki basit kalıbı izler -
Görüntülemek istediğiniz her bir ayrı özellik seti için bir Özellik Kümesi oluşturun ve bunlara Bio.SeqFeature nesneleri ekleyin.
Görüntülemek istediğiniz her grafik için bir Grafik Kümesi oluşturun ve bunlara grafik verileri ekleyin.
Diyagramda istediğiniz her parça için bir Parça oluşturun ve ihtiyacınız olan izlere Grafik Setleri ve Özellik Setleri ekleyin.
Bir Diyagram oluşturun ve İzleri buna ekleyin.
Diyagrama resmi çizmesini söyleyin.
Görüntüyü bir dosyaya yazın.
Giriş GenBank dosyasına bir örnek verelim -
https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbkve SeqRecord nesnesindeki kayıtları okuyun, ardından sonunda bir genom diyagramı çizin. Aşağıda açıklanmıştır,
Önce tüm modülleri aşağıda gösterildiği gibi ithal edeceğiz -
>>> from reportlab.lib import colors
>>> from reportlab.lib.units import cm
>>> from Bio.Graphics import GenomeDiagramŞimdi, verileri okumak için SeqIO modülünü içe aktarın -
>>> from Bio import SeqIO
record = SeqIO.read("example.gb", "genbank")Burada kayıt, diziyi genbank dosyasından okur.
Şimdi, parça ve özellik kümesi eklemek için boş bir diyagram oluşturun -
>>> diagram = GenomeDiagram.Diagram(
"Yersinia pestis biovar Microtus plasmid pPCP1")
>>> track = diagram.new_track(1, name="Annotated Features")
>>> feature = track.new_set()Şimdi, aşağıda tanımlandığı gibi yeşilden griye alternatif renkler kullanarak renk teması değişiklikleri uygulayabiliriz -
>>> for feature in record.features:
>>> if feature.type != "gene":
>>> continue
>>> if len(feature) % 2 == 0:
>>> color = colors.blue
>>> else:
>>> color = colors.red
>>>
>>> feature.add_feature(feature, color=color, label=True)Artık ekranınızda aşağıdaki yanıtı görebilirsiniz -
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dc90>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dfd0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x1007627d0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57290>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57050>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57390>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57590>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57410>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57490>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d574d0>Yukarıdaki girdi kayıtları için bir şema çizelim -
>>> diagram.draw(
format = "linear", orientation = "landscape", pagesize = 'A4',
... fragments = 4, start = 0, end = len(record))
>>> diagram.write("orchid.pdf", "PDF")
>>> diagram.write("orchid.eps", "EPS")
>>> diagram.write("orchid.svg", "SVG")
>>> diagram.write("orchid.png", "PNG")Yukarıdaki komutu çalıştırdıktan sonra, aşağıdaki görüntünün Biopython dizininize kaydedildiğini görebilirsiniz.
** Result **
genome.png
Aşağıdaki değişiklikleri yaparak resmi dairesel biçimde de çizebilirsiniz -
>>> diagram.draw(
format = "circular", circular = True, pagesize = (20*cm,20*cm),
... start = 0, end = len(record), circle_core = 0.7)
>>> diagram.write("circular.pdf", "PDF")Kromozomlara Genel Bakış
DNA molekülü, kromozom adı verilen iplik benzeri yapılar halinde paketlenir. Her kromozom, yapısını destekleyen histon adı verilen proteinlerin etrafına birçok kez sıkıca sarılmış DNA'dan oluşur.
Hücre bölünmediğinde, hücrenin çekirdeğinde kromozomlar - mikroskop altında bile - görünmez. Bununla birlikte, kromozomları oluşturan DNA, hücre bölünmesi sırasında daha sıkı bir şekilde paketlenir ve daha sonra bir mikroskop altında görünür hale gelir.
İnsanlarda, her hücre normalde toplam 46 olmak üzere 23 çift kromozom içerir. Otozom adı verilen bu çiftlerden yirmi ikisi, hem erkeklerde hem de dişilerde aynı görünür. 23. çift, cinsiyet kromozomları, erkekler ve kadınlar arasında farklılık gösterir. Dişilerde X kromozomunun iki kopyası varken erkeklerde bir X ve bir Y kromozomu vardır.
Fenotip, belirli bir kimyasala veya çevreye karşı bir organizma tarafından sergilenen gözlemlenebilir bir karakter veya özellik olarak tanımlanır. Fenotip mikrodizi aynı anda bir organizmanın çok sayıda kimyasal ve ortama karşı tepkisini ölçer ve gen mutasyonunu, gen karakterlerini vb. Anlamak için verileri analiz eder.
Biopython, fenotipik verileri analiz etmek için mükemmel bir Bio.Phenotype modülü sağlar. Bu bölümde fenotip mikroarray verilerinin nasıl ayrıştırılacağını, ara değerleneceğini, çıkarılacağını ve analiz edileceğini öğrenelim.
Ayrıştırma
Fenotip mikrodizi verileri iki formatta olabilir: CSV ve JSON. Biopython her iki formatı da destekler. Biopython ayrıştırıcı, fenotip mikroarray verilerini ayrıştırır ve PlateRecord nesnelerinin bir koleksiyonu olarak geri döner. Her PlateRecord nesnesi, WellRecord nesnelerinin bir koleksiyonunu içerir. Her WellRecord nesnesi, verileri 8 satır ve 12 sütun biçiminde tutar. Sekiz sıranın H ile temsil edilmektedir ve 12 sütun Örnek 12 ', 4 01 ile temsil edilmektedir inci satır ve 6 inci D06 ile temsil sütununda gösterilmiştir.
Ayrıştırma biçimini ve kavramını aşağıdaki örnekle anlayalım -
Step 1 - Biopython ekibi tarafından sağlanan Plates.csv dosyasını indirin - https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/Plates.csv
Step 2 - Fenotp modülünü aşağıdaki gibi yükleyin -
>>> from Bio import phenotypeStep 3- Veri dosyasını ve biçim seçeneğini ("pm-csv") geçirerek phenotype.parse yöntemini çağırın. Yinelenebilir PlateRecord'u aşağıdaki gibi döndürür,
>>> plates = list(phenotype.parse('Plates.csv', "pm-csv"))
>>> plates
[PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'],WellRecord['A03'], ..., WellRecord['H12']')]
>>>Step 4 - Aşağıdaki listeden ilk plakaya erişin -
>>> plate = plates[0]
>>> plate
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ...,
WellRecord['H12']')
>>>Step 5- Daha önce tartışıldığı gibi, bir tabak her biri 12 maddeden oluşan 8 sıra içerir. WellRecord'a aşağıda belirtildiği gibi iki şekilde erişilebilir -
>>> well = plate["A04"]
>>> well = plate[0, 4]
>>> well WellRecord('(0.0, 0.0), (0.25, 0.0), (0.5, 0.0), (0.75, 0.0),
(1.0, 0.0), ..., (71.75, 388.0)')
>>>Step 6 - Her kuyucuğun farklı zaman noktalarında bir dizi ölçümü olacaktır ve aşağıda belirtildiği gibi for döngüsü kullanılarak erişilebilir -
>>> for v1, v2 in well:
... print(v1, v2)
...
0.0 0.0
0.25 0.0
0.5 0.0
0.75 0.0
1.0 0.0
...
71.25 388.0
71.5 388.0
71.75 388.0
>>>İnterpolasyon
Enterpolasyon, verilere daha fazla içgörü sağlar. Biopython, ara zaman noktaları için bilgi almak üzere WellRecord verilerinin enterpolasyonuna yönelik yöntemler sağlar. Sözdizimi liste indekslemeye benzer ve bu yüzden öğrenmesi kolaydır.
Verileri 20.1 saatte almak için, aşağıda belirtildiği gibi dizin değerleri olarak geçirmeniz yeterlidir -
>>> well[20.10]
69.40000000000003
>>>Başlangıç ve bitiş zaman noktalarının yanı sıra aşağıda belirtildiği gibi geçebiliriz -
>>> well[20:30]
[67.0, 84.0, 102.0, 119.0, 135.0, 147.0, 158.0, 168.0, 179.0, 186.0]
>>>Yukarıdaki komut, 1 saatlik aralıklarla 20 saatten 30 saate kadar verilerin enterpolasyonunu yapar. Varsayılan olarak, aralık 1 saattir ve herhangi bir değere değiştirebiliriz. Örneğin, aşağıda belirtildiği gibi 15 dakikalık (0.25 saat) bir aralık verelim -
>>> well[20:21:0.25]
[67.0, 73.0, 75.0, 81.0]
>>>Analiz Et ve Çıkar
Biopython, Gompertz, Logistic ve Richards sigmoid fonksiyonlarını kullanarak WellRecord verilerini analiz etmek için uygun bir yöntem sağlar. Varsayılan olarak, fit yöntemi Gompertz işlevini kullanır. Görevi tamamlamak için WellRecord nesnesinin fit yöntemini çağırmamız gerekir. Kodlama aşağıdaki gibidir -
>>> well.fit()
Traceback (most recent call last):
...
Bio.MissingPythonDependencyError: Install scipy to extract curve parameters.
>>> well.model
>>> getattr(well, 'min') 0.0
>>> getattr(well, 'max') 388.0
>>> getattr(well, 'average_height')
205.42708333333334
>>>Biopython, gelişmiş analiz yapmak için scipy modülüne bağlıdır. Scipy modülünü kullanmadan min, max ve average_height detaylarını hesaplayacaktır.
Bu bölüm sekansların nasıl çizileceğini açıklar. Bu konuya geçmeden önce, çizim yapmanın temellerini anlayalım.
Çizim
Matplotlib, çeşitli formatlarda kaliteli rakamlar üreten bir Python çizim kitaplığıdır. Çizgi grafik, histogramlar, çubuk grafik, pasta grafik, dağılım grafiği vb. Gibi farklı grafik türleri oluşturabiliriz.
pyLab is a module that belongs to the matplotlib which combines the numerical module numpy with the graphical plotting module pyplot.Biopython, dizileri çizmek için pylab modülünü kullanır. Bunu yapmak için aşağıdaki kodu içe aktarmamız gerekiyor -
import pylabİçe aktarmadan önce, matplotlib paketini aşağıdaki komutla pip komutunu kullanarak kurmamız gerekir -
pip install matplotlibÖrnek Giriş Dosyası
Adlı bir örnek dosya oluşturun plot.fasta Biopython dizininizde ve aşağıdaki değişiklikleri ekleyin -
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF
>seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME
>seq2 EEYQTWEEFARAAEKLYLTDPMKVRVVLKYRHCDGNLCMKVTDDAVCLQYKTDQAQDVKKVEKLHGK
>seq3 MYQVWEEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVCLQYKTDQAQDV
>seq4 EEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVVSYEMRLFGVQKDNFALEHSLL
>seq5 SWEEFAKAAEVLYLEDPMKCRMCTKYRHVDHKLVVKLTDNHTVLKYVTDMAQDVKKIEKLTTLLMR
>seq6 FTNWEEFAKAAERLHSANPEKCRFVTKYNHTKGELVLKLTDDVVCLQYSTNQLQDVKKLEKLSSTLLRSI
>seq7 SWEEFVERSVQLFRGDPNATRYVMKYRHCEGKLVLKVTDDRECLKFKTDQAQDAKKMEKLNNIFF
>seq8 SWDEFVDRSVQLFRADPESTRYVMKYRHCDGKLVLKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM
>seq9 KNWEDFEIAAENMYMANPQNCRYTMKYVHSKGHILLKMSDNVKCVQYRAENMPDLKK
>seq10 FDSWDEFVSKSVELFRNHPDTTRYVVKYRHCEGKLVLKVTDNHECLKFKTDQAQDAKKMEKÇizgi Grafiği
Şimdi, yukarıdaki fasta dosyası için basit bir çizgi grafiği oluşturalım.
Step 1 - fasta dosyasını okumak için SeqIO modülünü içe aktarın.
>>> from Bio import SeqIOStep 2 - Girdi dosyasını ayrıştırın.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Pilab modülünü ithal edelim.
>>> import pylabStep 4 - x ve y ekseni etiketleri atayarak çizgi grafiğini yapılandırın.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Izgara görünümünü ayarlayarak çizgi grafiğini yapılandırın.
>>> pylab.grid()Step 6 - Plot yöntemini çağırarak ve kayıtları girdi olarak sağlayarak basit çizgi grafiği çizin.
>>> pylab.plot(records)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Son olarak aşağıdaki komutu kullanarak grafiği kaydedin.
>>> pylab.savefig("lines.png")Sonuç
Yukarıdaki komutu çalıştırdıktan sonra, aşağıdaki görüntünün Biopython dizininize kaydedildiğini görebilirsiniz.
Histogram Grafiği
Bölmelerin veri aralıklarını temsil ettiği sürekli veriler için bir histogram kullanılır. Çizim histogramı, pylab.plot dışında çizgi grafikle aynıdır. Bunun yerine, kayıtlarla pilab modülünün hist yöntemini ve kutular için bazı custum değerini çağırın (5). Tam kodlama aşağıdaki gibidir -
Step 1 - fasta dosyasını okumak için SeqIO modülünü içe aktarın.
>>> from Bio import SeqIOStep 2 - Girdi dosyasını ayrıştırın.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Pilab modülünü ithal edelim.
>>> import pylabStep 4 - x ve y ekseni etiketleri atayarak çizgi grafiğini yapılandırın.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Izgara görünümünü ayarlayarak çizgi grafiğini yapılandırın.
>>> pylab.grid()Step 6 - Plot yöntemini çağırarak ve kayıtları girdi olarak sağlayarak basit çizgi grafiği çizin.
>>> pylab.hist(records,bins=5)
(array([2., 3., 1., 3., 2.]), array([57., 60., 63., 66., 69., 72.]), <a list
of 5 Patch objects>)
>>>Step 7 - Son olarak aşağıdaki komutu kullanarak grafiği kaydedin.
>>> pylab.savefig("hist.png")Sonuç
Yukarıdaki komutu çalıştırdıktan sonra, aşağıdaki görüntünün Biopython dizininize kaydedildiğini görebilirsiniz.
Sıradaki GC Yüzdesi
GC yüzdesi, farklı dizileri karşılaştırmak için yaygın olarak kullanılan analitik verilerden biridir. Bir dizi dizinin GC Yüzdesini kullanarak basit bir çizgi grafik oluşturabilir ve hemen karşılaştırabiliriz. Burada, verileri sıra uzunluğundan GC yüzdesine değiştirebiliriz. Tam kodlama aşağıda verilmiştir -
Step 1 - fasta dosyasını okumak için SeqIO modülünü içe aktarın.
>>> from Bio import SeqIOStep 2 - Girdi dosyasını ayrıştırın.
>>> from Bio.SeqUtils import GC
>>> gc = sorted(GC(rec.seq) for rec in SeqIO.parse("plot.fasta", "fasta"))Step 3 - Pilab modülünü ithal edelim.
>>> import pylabStep 4 - x ve y ekseni etiketleri atayarak çizgi grafiğini yapılandırın.
>>> pylab.xlabel("Genes")
Text(0.5, 0, 'Genes')
>>> pylab.ylabel("GC Percentage")
Text(0, 0.5, 'GC Percentage')
>>>Step 5 - Izgara görünümünü ayarlayarak çizgi grafiğini yapılandırın.
>>> pylab.grid()Step 6 - Plot yöntemini çağırarak ve kayıtları girdi olarak sağlayarak basit çizgi grafiği çizin.
>>> pylab.plot(gc)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Son olarak aşağıdaki komutu kullanarak grafiği kaydedin.
>>> pylab.savefig("gc.png")Sonuç
Yukarıdaki komutu çalıştırdıktan sonra, aşağıdaki görüntünün Biopython dizininize kaydedildiğini görebilirsiniz.
Genel olarak, Küme analizi, aynı gruptaki bir dizi nesneyi gruplamaktır. Bu kavram temel olarak veri madenciliği, istatistiksel veri analizi, makine öğrenimi, örüntü tanıma, görüntü analizi, biyoinformatik vb. Alanlarda kullanılır. Kümenin farklı analizlerde yaygın olarak nasıl kullanıldığını anlamak için çeşitli algoritmalarla elde edilebilir.
Bioinformatics'e göre, küme analizi, benzer gen ifadesine sahip gen gruplarını bulmak için esas olarak gen ekspresyon veri analizinde kullanılır.
Bu bölümde, gerçek bir veri setinde kümelemenin temellerini anlamak için Biopython'daki önemli algoritmaları inceleyeceğiz.
Biopython, tüm algoritmaları uygulamak için Bio.Cluster modülünü kullanır. Aşağıdaki algoritmaları destekler -
- Hiyerarşik kümeleme
- K - Kümeleme
- Kendi Kendini Düzenleyen Haritalar
- Temel bileşenler Analizi
Yukarıdaki algoritmalara kısa bir giriş yapalım.
Hiyerarşik kümeleme
Hiyerarşik kümeleme, her düğümü bir uzaklık ölçüsü ile en yakın komşusuna bağlamak ve bir küme oluşturmak için kullanılır. Bio.Cluster düğümünün üç özelliği vardır: sol, sağ ve mesafe. Aşağıda gösterildiği gibi basit bir küme oluşturalım -
>>> from Bio.Cluster import Node
>>> n = Node(1,10)
>>> n.left = 11
>>> n.right = 0
>>> n.distance = 1
>>> print(n)
(11, 0): 1Ağaç tabanlı kümeleme oluşturmak istiyorsanız, aşağıdaki komutu kullanın -
>>> n1 = [Node(1, 2, 0.2), Node(0, -1, 0.5)] >>> n1_tree = Tree(n1)
>>> print(n1_tree)
(1, 2): 0.2
(0, -1): 0.5
>>> print(n1_tree[0])
(1, 2): 0.2Bio.Cluster modülünü kullanarak hiyerarşik kümeleme yapalım.
Mesafenin bir dizide tanımlandığını düşünün.
>>> import numpy as np
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Şimdi uzaklık dizisini ağaç kümesine ekleyin.
>>> from Bio.Cluster import treecluster
>>> cluster = treecluster(distance)
>>> print(cluster)
(2, 1): 0.666667
(-1, 0): 9.66667Yukarıdaki işlev bir Ağaç küme nesnesi döndürür. Bu nesne, öğe sayısının satırlar veya sütunlar olarak kümelendiği düğümleri içerir.
K - Kümeleme
Bu bir tür bölümleme algoritmasıdır ve k - ortalamalar, medyanlar ve medoid kümelemesi olarak sınıflandırılır. Her bir kümelenmeyi kısaca anlayalım.
K-anlamına gelir Kümeleme
Bu yaklaşım veri madenciliğinde popülerdir. Bu algoritmanın amacı, K değişkeni ile temsil edilen grup sayısı ile verilerdeki grupları bulmaktır.
Algoritma, her veri noktasını sağlanan özelliklere bağlı olarak K gruplarından birine atamak için yinelemeli olarak çalışır. Veri noktaları, özellik benzerliğine göre kümelenmiştir.
>>> from Bio.Cluster import kcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid, error,found = kcluster(data)
>>> print(clusterid) [0 0 1]
>>> print(found)
1K-medyan Kümeleme
Merkezini belirlemek için her kümenin ortalamasını hesaplayan başka bir tür kümeleme algoritmasıdır.
K-medoids Kümeleme
Bu yaklaşım, mesafe matrisini ve kullanıcı tarafından geçen küme sayısını kullanan belirli bir dizi maddeye dayanmaktadır.
Mesafe matrisini aşağıda tanımlandığı gibi düşünün -
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Aşağıdaki komutu kullanarak k-medoids kümelemesini hesaplayabiliriz -
>>> from Bio.Cluster import kmedoids
>>> clusterid, error, found = kmedoids(distance)Bir örnek düşünelim.
Kcluster işlevi, Seq örneklerini değil girdi olarak bir veri matrisini alır. Dizilerinizi bir matrise dönüştürmeniz ve bunu kcluster işlevine sağlamanız gerekir.
Verileri yalnızca sayısal öğeler içeren bir matrise dönüştürmenin bir yolu, numpy.fromstringişlevi. Temel olarak, bir sıradaki her harfi ASCII karşılığına çevirir.
Bu, kcluster işlevinin tanıdığı ve dizilerinizi kümelemek için kullandığı 2D kodlanmış diziler dizisi oluşturur.
>>> from Bio.Cluster import kcluster
>>> import numpy as np
>>> sequence = [ 'AGCT','CGTA','AAGT','TCCG']
>>> matrix = np.asarray([np.fromstring(s, dtype=np.uint8) for s in sequence])
>>> clusterid,error,found = kcluster(matrix)
>>> print(clusterid) [1 0 0 1]Kendi Kendini Düzenleyen Haritalar
Bu yaklaşım bir tür yapay sinir ağıdır. Kohonen tarafından geliştirilmiştir ve genellikle Kohonen haritası olarak adlandırılır. Öğeleri dikdörtgen topolojiye göre kümeler halinde düzenler.
Aşağıda gösterildiği gibi aynı dizi mesafesini kullanarak basit bir küme oluşturalım -
>>> from Bio.Cluster import somcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid,map = somcluster(data)
>>> print(map)
[[[-1.36032469 0.38667395]]
[[-0.41170578 1.35295911]]]
>>> print(clusterid)
[[1 0]
[1 0]
[1 0]]Buraya, clusterid satır sayısının kümelenmiş öğelerin sayısına eşit olduğu iki sütunlu bir dizidir ve data satırlar veya sütunlar boyutlarına sahip bir dizidir.
Temel bileşenler Analizi
Temel Bileşen Analizi, yüksek boyutlu verileri görselleştirmek için kullanışlıdır. Orijinal verilerin aynı sayıda veya daha az boyuta projeksiyonunu hesaplamak için doğrusal cebirden ve istatistikten basit matris işlemlerini kullanan bir yöntemdir.
Temel Bileşen Analizi bir demet sütun ortalamasını, koordinatları, bileşenleri ve özdeğerleri döndürür. Bu kavramın temellerine bakalım.
>>> from numpy import array
>>> from numpy import mean
>>> from numpy import cov
>>> from numpy.linalg import eig
# define a matrix
>>> A = array([[1, 2], [3, 4], [5, 6]])
>>> print(A)
[[1 2]
[3 4]
[5 6]]
# calculate the mean of each column
>>> M = mean(A.T, axis = 1)
>>> print(M)
[ 3. 4.]
# center columns by subtracting column means
>>> C = A - M
>>> print(C)
[[-2. -2.]
[ 0. 0.]
[ 2. 2.]]
# calculate covariance matrix of centered matrix
>>> V = cov(C.T)
>>> print(V)
[[ 4. 4.]
[ 4. 4.]]
# eigendecomposition of covariance matrix
>>> values, vectors = eig(V)
>>> print(vectors)
[[ 0.70710678 -0.70710678]
[ 0.70710678 0.70710678]]
>>> print(values)
[ 8. 0.]Aşağıda tanımlanan aynı dikdörtgen matris verilerini Bio.Cluster modülüne uygulayalım -
>>> from Bio.Cluster import pca
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> columnmean, coordinates, components, eigenvalues = pca(data)
>>> print(columnmean)
[ 3. 4.]
>>> print(coordinates)
[[-2.82842712 0. ]
[ 0. 0. ]
[ 2.82842712 0. ]]
>>> print(components)
[[ 0.70710678 0.70710678]
[ 0.70710678 -0.70710678]]
>>> print(eigenvalues)
[ 4. 0.]Biyoinformatik, makine öğrenimi algoritmalarını uygulamak için mükemmel bir alandır. Burada çok sayıda organizmanın genetik bilgisine sahibiz ve tüm bu bilgileri manuel olarak analiz etmek mümkün değil. Uygun makine öğrenimi algoritması kullanılırsa, bu verilerden pek çok yararlı bilgi çıkarabiliriz. Biopython, denetimli makine öğrenimi yapmak için yararlı bir algoritma seti sağlar.
Denetimli öğrenme, girdi değişkenine (X) ve çıktı değişkenine (Y) dayanır. Girişten çıkışa eşleme işlevini öğrenmek için bir algoritma kullanır. Aşağıda tanımlanmıştır -
Y = f(X)Bu yaklaşımın temel amacı, eşleme işlevine yaklaşmaktır ve yeni girdi verileriniz (x) olduğunda, bu veriler için çıktı değişkenlerini (Y) tahmin edebilirsiniz.
Lojistik Regresyon Modeli
Lojistik regresyon, denetlenen bir makine öğrenimi algoritmasıdır. Yordayıcı değişkenlerin ağırlıklı toplamını kullanarak K sınıfları arasındaki farkı bulmak için kullanılır. Bir olayın meydana gelme olasılığını hesaplar ve kanser tespiti için kullanılabilir.
Biopython, Lojistik regresyon algoritmasına dayalı olarak değişkenleri tahmin etmek için Bio.LogisticRegression modülü sağlar. Şu anda Biopython, yalnızca iki sınıf için lojistik regresyon algoritması uygulamaktadır (K = 2).
k-En Yakın Komşular
k-En yakın komşular aynı zamanda denetimli bir makine öğrenimi algoritmasıdır. Verileri en yakın komşulara göre kategorize ederek çalışır. Biopython, değişkenleri k en yakın komşu algoritmasına göre tahmin etmek için Bio.KNN modülü sağlar.
Naif bayanlar
Naive Bayes sınıflandırıcıları, Bayes Teoremine dayalı bir sınıflandırma algoritmaları koleksiyonudur. Tek bir algoritma değil, hepsinin ortak bir ilkeyi paylaştığı bir algoritma ailesidir, yani sınıflandırılan her özellik çifti birbirinden bağımsızdır. Biopython, Naive Bayes algoritmasıyla çalışmak için Bio.NaiveBayes modülü sağlar.
Markov Modeli
Bir Markov modeli, belirli olasılık kurallarına göre bir durumdan diğerine geçişi deneyimleyen rastgele değişkenlerin bir koleksiyonu olarak tanımlanan matematiksel bir sistemdir. Biopython sağlarBio.MarkovModel and Bio.HMM.MarkovModel modules to work with Markov models.
Biopython, yazılımın hatasız olduğundan emin olmak için yazılımı farklı koşullar altında test etmek için kapsamlı bir test komut dosyasına sahiptir. Test komut dosyasını çalıştırmak için Biopython'un kaynak kodunu indirin ve ardından aşağıdaki komutu çalıştırın -
python run_tests.pyBu, tüm test komut dosyalarını çalıştıracak ve aşağıdaki çıktıyı verecektir -
Python version: 2.7.12 (v2.7.12:d33e0cf91556, Jun 26 2016, 12:10:39)
[GCC 4.2.1 (Apple Inc. build 5666) (dot 3)]
Operating system: posix darwin
test_Ace ... ok
test_Affy ... ok
test_AlignIO ... ok
test_AlignIO_ClustalIO ... ok
test_AlignIO_EmbossIO ... ok
test_AlignIO_FastaIO ... ok
test_AlignIO_MauveIO ... ok
test_AlignIO_PhylipIO ... ok
test_AlignIO_convert ... ok
...........................................
...........................................Ayrıca aşağıda belirtildiği gibi ayrı test komut dosyası çalıştırabiliriz -
python test_AlignIO.pySonuç
Biopython, öğrendiğimiz gibi, biyoinformatik alanındaki önemli yazılımlardan biridir. Python ile yazılmış (öğrenmesi ve yazması kolay), biyoinformatik alanındaki herhangi bir hesaplama ve işlemle başa çıkmak için kapsamlı işlevsellik sağlar. Ayrıca işlevselliğinden yararlanmak için neredeyse tüm popüler biyoinformatik yazılımlarına kolay ve esnek bir arayüz sağlar.