Biopython - moduł BioSQL
BioSQLto ogólny schemat bazy danych przeznaczony głównie do przechowywania sekwencji i powiązanych z nimi danych dla wszystkich silników RDBMS. Został zaprojektowany w taki sposób, że przechowuje dane ze wszystkich popularnych baz danych bioinformatycznych, takich jak GenBank, Swissport itp. Może być również używany do przechowywania danych wewnętrznych.
BioSQL obecnie zapewnia określony schemat dla poniższych baz danych -
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Zapewnia również minimalne wsparcie dla baz danych HSQLDB i Derby opartych na języku Java.
BioPython zapewnia bardzo proste, łatwe i zaawansowane możliwości ORM do pracy z bazą danych opartą na BioSQL. BioPython provides a module, BioSQL wykonać następującą funkcjonalność -
- Utwórz / usuń bazę danych BioSQL
- Połącz się z bazą danych BioSQL
- Przeanalizuj bazę danych sekwencji, taką jak GenBank, Swisport, wynik BLAST, wynik Entrez itp., I załaduj ją bezpośrednio do bazy danych BioSQL
- Pobierz dane sekwencji z bazy danych BioSQL
- Pobierz dane taksonomii z NCBI BLAST i przechowuj je w bazie danych BioSQL
- Uruchom dowolne zapytanie SQL w bazie danych BioSQL
Omówienie schematu bazy danych BioSQL
Zanim zagłębimy się w BioSQL, pozwól nam zrozumieć podstawy schematu BioSQL. Schemat BioSQL zapewnia ponad 25 tabel do przechowywania danych sekwencji, cech sekwencji, kategorii / ontologii sekwencji i informacji taksonomicznych. Oto niektóre z ważnych tabel -
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
Tworzenie bazy danych BioSQL
W tej sekcji stwórzmy przykładową bazę danych BioSQL, biosql przy użyciu schematu dostarczonego przez zespół BioSQL. Będziemy pracować z bazą danych SQLite, ponieważ jest naprawdę łatwa do rozpoczęcia i nie ma skomplikowanej konfiguracji.
W tym miejscu utworzymy bazę danych BioSQL opartą na SQLite, wykonując poniższe kroki.
Step 1 - Pobierz silnik bazy danych SQLite i zainstaluj go.
Step 2 - Pobierz projekt BioSQL z adresu URL GitHub. https://github.com/biosql/biosql
Step 3 - Otwórz konsolę i utwórz katalog za pomocą mkdir i wejdź do niego.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 - Uruchom poniższe polecenie, aby utworzyć nową bazę danych SQLite.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 - Skopiuj plik biosqldb-sqlite.sql z projektu BioSQL (/ sql / biosqldb-sqlite.sql`) i zapisz go w bieżącym katalogu.
Step 6 - Uruchom poniższe polecenie, aby utworzyć wszystkie tabele.
sqlite> .read biosqldb-sqlite.sqlTeraz wszystkie tabele są tworzone w naszej nowej bazie danych.
Step 7 - Uruchom poniższe polecenie, aby wyświetlić wszystkie nowe tabele w naszej bazie danych.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>Pierwsze trzy polecenia to polecenia konfiguracyjne służące do skonfigurowania programu SQLite w celu wyświetlenia wyniku w sformatowany sposób.
Step 8 - Skopiuj przykładowy plik GenBank, ls_orchid.gbk dostarczony przez zespół BioPython https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk do bieżącego katalogu i zapisz go jako orchid.gbk.
Step 9 - Utwórz skrypt Pythona, load_orchid.py, używając poniższego kodu i wykonaj go.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Powyższy kod analizuje rekord w pliku i konwertuje go na obiekty Pythona i wstawia do bazy danych BioSQL. Przeanalizujemy kod w dalszej części.
Na koniec stworzyliśmy nową bazę danych BioSQL i załadowaliśmy do niej kilka przykładowych danych. W następnym rozdziale omówimy ważne tabele.
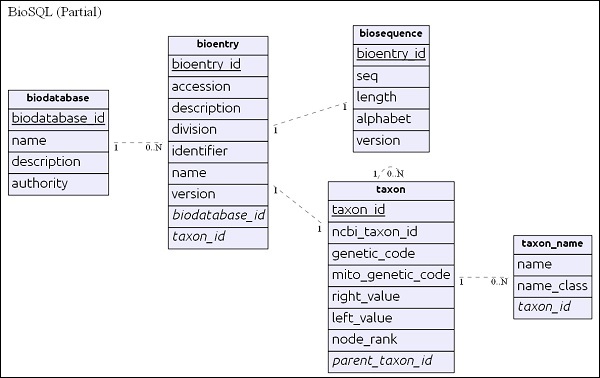
Prosty schemat ER
biodatabase tabela znajduje się na szczycie hierarchii, a jej głównym celem jest zorganizowanie zestawu danych sekwencyjnych w pojedynczą grupę / wirtualną bazę danych. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. Wszystkie powiązane tabele w bazie danych BioSQL zawierają odniesienia do wpisu bazy danych biologicznych.
bioentrytabela zawiera wszystkie szczegóły dotyczące sekwencji z wyjątkiem danych sekwencji. dane sekwencyjne konkretnegobioentry będą przechowywane w biosequence stół.
Takson i nazwa_takonu są szczegółami taksonomii i każda pozycja odsyła do tej tabeli w celu określenia informacji o taksonu.
Po zrozumieniu schematu przyjrzyjmy się niektórym zapytaniom w następnej sekcji.
Zapytania BioSQL
Zagłębmy się w niektóre zapytania SQL, aby lepiej zrozumieć, w jaki sposób dane są zorganizowane i tabele są ze sobą powiązane. Zanim przejdziesz dalej, otwórzmy bazę danych za pomocą poniższego polecenia i ustaw kilka poleceń formatujących -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Możesz również użyć dowolnego edytora SQLite, aby uruchomić zapytanie.
Wymień wirtualną bazę danych sekwencji dostępną w systemie, jak podano poniżej -
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Tutaj mamy tylko jedną bazę danych, orchid.
Wypisz wpisy (pierwsze 3) dostępne w bazie danych orchid z poniższym kodem
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Wymień szczegóły sekwencji związane z wpisem (przystąpienie - Z78530, nazwa - gen C. fasciculatum 5.8S rRNA oraz DNA ITS1 i ITS2) z podanym kodem -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Uzyskaj pełną sekwencję związaną z wpisem (dostęp - Z78530, nazwa - gen C. fasciculatum 5.8S rRNA oraz DNA ITS1 i ITS2), używając poniższego kodu -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Lista taksonów powiązanych z bio bazą danych, orchidea
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>Załaduj dane do bazy danych BioSQL
W tym rozdziale nauczmy się, jak ładować dane sekwencji do bazy danych BioSQL. Mamy już kod do ładowania danych do bazy danych w poprzedniej sekcji, a kod wygląda następująco -
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Przyjrzymy się dokładniej każdej linii kodu i jego przeznaczeniu -
Line 1 - Ładuje moduł SeqIO.
Line 2- Ładuje moduł BioSeqDatabase. Ten moduł zapewnia wszystkie funkcje do interakcji z bazą danych BioSQL.
Line 3 - Ładuje moduł systemu operacyjnego.
Line 5- open_database otwiera określoną bazę danych (db) ze skonfigurowanym sterownikiem (sterownikiem) i zwraca uchwyt do bazy danych BioSQL (serwera). Biopython obsługuje bazy danych sqlite, mysql, postgresql i oracle.
Line 6-10- metoda load_database_sql ładuje sql z zewnętrznego pliku i wykonuje go. Metoda commit zatwierdza transakcję. Możemy pominąć ten krok, ponieważ utworzyliśmy już bazę danych ze schematem.
Line 12 - metody new_database tworzy nową wirtualną bazę danych, orchid i zwraca db uchwyt do wykonania polecenia w bazie danych orchidei.
Line 13- metoda load ładuje wpisy sekwencji (iterowalny SeqRecord) do bazy danych orchidei. SqlIO.parse analizuje bazę danych GenBank i zwraca wszystkie zawarte w niej sekwencje jako iterowalne SeqRecord. Drugi parametr (True) metody load nakazuje jej pobranie szczegółów taksonomii danych sekwencji ze strony internetowej NCBI, jeśli nie są one jeszcze dostępne w systemie.
Line 14 - commit zatwierdza transakcję.
Line 15 - close zamyka połączenie z bazą danych i niszczy uchwyt serwera.
Pobierz dane sekwencji
Pobierzmy sekwencję z identyfikatorem 2765658 z bazy danych orchidei, jak poniżej -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Tutaj serwer ["orchid"] zwraca uchwyt do pobierania danych z wirtualnej bazy danychorchid. lookup metoda zapewnia opcję wyboru sekwencji na podstawie kryteriów i wybraliśmy sekwencję o identyfikatorze 2765658. lookupzwraca informacje o sekwencji jako SeqRecordobject. Ponieważ wiemy już, jak pracować z SeqRecord`, łatwo jest uzyskać z niego dane.
Usuń bazę danych
Usunięcie bazy danych jest tak proste, jak wywołanie metody remove_database z odpowiednią nazwą bazy danych, a następnie zatwierdzenie jej zgodnie z poniższym opisem -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()