Biopython - Szybki przewodnik
Biopython to największy i najpopularniejszy pakiet bioinformatyczny dla Pythona. Zawiera szereg różnych podmodułów do typowych zadań bioinformatycznych. Został opracowany przez Chapmana i Changa, głównie w języku Python. Zawiera również kod C do optymalizacji złożonej części obliczeniowej oprogramowania. Działa w systemach Windows, Linux, Mac OS X itp.
Zasadniczo Biopython to zbiór modułów Pythona, które zapewniają funkcje do obsługi sekwencji DNA, RNA i białek, takich jak odwrotne uzupełnianie ciągu DNA, znajdowanie motywów w sekwencjach białek itp. Zapewnia wiele parserów do odczytu wszystkich głównych genetycznych baz danych takich jak GenBank, SwissPort, FASTA itp., a także otoki / interfejsy do uruchamiania innych popularnych programów / narzędzi bioinformatycznych, takich jak NCBI BLASTN, Entrez itp., w środowisku Pythona. Ma podobne projekty, takie jak BioPerl, BioJava i BioRuby.
funkcje
Biopython jest przenośny, przejrzysty i ma łatwą do nauczenia składnię. Poniżej wymieniono niektóre z najważniejszych funkcji -
Interpretowane, interaktywne i obiektowe.
Obsługuje formaty powiązane z FASTA, PDB, GenBank, Blast, SCOP, PubMed / Medline, ExPASy.
Możliwość radzenia sobie z formatami sekwencji.
Narzędzia do zarządzania strukturami białek.
BioSQL - standardowy zestaw tabel SQL do przechowywania sekwencji oraz funkcji i adnotacji.
Dostęp do usług online i baz danych, w tym usług NCBI (Blast, Entrez, PubMed) i usług ExPASY (SwissProt, Prosite).
Dostęp do usług lokalnych, w tym Blast, Clustalw, EMBOSS.
Cele
Celem Biopython jest zapewnienie prostego, standardowego i szerokiego dostępu do bioinformatyki za pośrednictwem języka Python. Konkretne cele Biopythonu są wymienione poniżej -
Zapewnienie ustandaryzowanego dostępu do zasobów bioinformatycznych.
Wysokiej jakości moduły i skrypty wielokrotnego użytku.
Szybka manipulacja tablicą, której można używać w kodzie klastra, PDB, NaiveBayes i modelu Markov.
Analiza danych genomowych.
Zalety
Biopython wymaga bardzo mniej kodu i ma następujące zalety -
Zapewnia typ danych mikromacierzy używany w klastrowaniu.
Odczytuje i zapisuje pliki typu Tree-View.
Obsługuje dane strukturalne używane do analizowania, reprezentacji i analizy PDB.
Obsługuje dane dziennika używane w aplikacjach Medline.
Obsługuje bazę danych BioSQL, która jest powszechnie używaną standardową bazą danych we wszystkich projektach bioinformatycznych.
Obsługuje rozwój parsera, dostarczając moduły do analizowania pliku bioinformatycznego do obiektu rekordu określonego formatu lub ogólnej klasy sekwencji i funkcji.
Przejrzysta dokumentacja oparta na stylu książki kucharskiej.
Przykładowe studium przypadku
Sprawdźmy niektóre przypadki użycia (genetyka populacji, struktura RNA itp.) I spróbujmy zrozumieć, jak Biopython odgrywa ważną rolę w tej dziedzinie -
Genetyka populacji
Genetyka populacji to badanie zmienności genetycznej w populacji i obejmuje badanie i modelowanie zmian w częstości występowania genów i alleli w populacji w czasie i przestrzeni.
Biopython dostarcza moduł Bio.PopGen do genetyki populacyjnej. Ten moduł zawiera wszystkie niezbędne funkcje do zbierania informacji o klasycznej genetyce populacji.
Struktura RNA
Trzy główne makrocząsteczki biologiczne, które są niezbędne dla naszego życia to DNA, RNA i Białko. Białka są końmi roboczymi komórki i odgrywają ważną rolę jako enzymy. DNA (kwas dezoksyrybonukleinowy) jest uważany za „plan” komórki. Zawiera wszystkie informacje genetyczne potrzebne komórce do wzrostu, pobierania składników odżywczych i rozmnażania. RNA (kwas rybonukleinowy) działa jako „fotokopia DNA” w komórce.
Biopython dostarcza obiekty Bio.Sequence, które reprezentują nukleotydy, elementy składowe DNA i RNA.
Ta sekcja wyjaśnia, jak zainstalować Biopython na twoim komputerze. Jest bardzo łatwy w instalacji i nie zajmie więcej niż pięć minut.
Step 1 - Weryfikacja instalacji Pythona
Biopython został zaprojektowany do pracy z wersjami Python 2.5 lub nowszymi. Dlatego konieczne jest, aby najpierw zainstalować Pythona. Uruchom poniższe polecenie w wierszu polecenia -
> python --versionJest zdefiniowany poniżej -
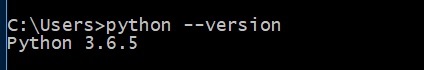
Pokazuje wersję Pythona, jeśli została poprawnie zainstalowana. W przeciwnym razie pobierz najnowszą wersję języka Python, zainstaluj ją, a następnie ponownie uruchom polecenie.
Step 2 - Instalacja Biopythona za pomocą pip
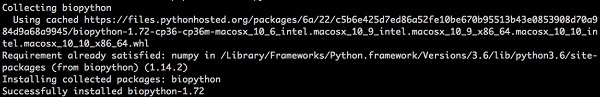
Biopython jest łatwy do zainstalowania przy użyciu pip z wiersza poleceń na wszystkich platformach. Wpisz poniższe polecenie -
> pip install biopythonNa ekranie pojawi się następująca odpowiedź -
Aby zaktualizować starszą wersję Biopythona -
> pip install biopython –-upgradeNa ekranie pojawi się następująca odpowiedź -
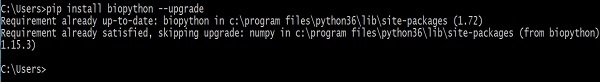
Po wykonaniu tego polecenia starsze wersje Biopython i NumPy (Biopython od tego zależy) zostaną usunięte przed zainstalowaniem ostatnich wersji.
Step 3 - Weryfikacja instalacji Biopythona
Teraz pomyślnie zainstalowałeś Biopython na swoim komputerze. Aby sprawdzić, czy Biopython jest poprawnie zainstalowany, wpisz poniższe polecenie na konsoli Pythona -
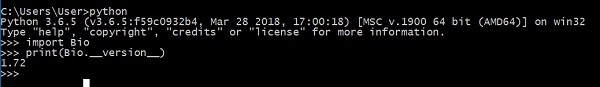
Pokazuje wersję Biopythona.
Alternate Way − Installing Biopython using Source
Aby zainstalować Biopython przy użyciu kodu źródłowego, postępuj zgodnie z poniższymi instrukcjami -
Pobierz najnowsze wydanie Biopython z poniższego linku - https://biopython.org/wiki/Download
Na razie najnowsza wersja to biopython-1.72.
Pobierz plik i rozpakuj skompresowany plik archiwum, przejdź do folderu kodu źródłowego i wpisz poniższe polecenie -
> python setup.py buildSpowoduje to zbudowanie Biopythona z kodu źródłowego, jak podano poniżej -
Teraz przetestuj kod za pomocą poniższego polecenia -
> python setup.py test
Na koniec zainstaluj za pomocą poniższego polecenia -
> python setup.py install
Stwórzmy prostą aplikację Biopython, aby przeanalizować plik bioinformatyki i wydrukować zawartość. Pomoże nam to zrozumieć ogólną koncepcję Biopythonu i tego, jak pomaga w dziedzinie bioinformatyki.
Step 1 - Najpierw utwórz przykładowy plik sekwencji „przyklad.fasta” i umieść w nim poniższą zawartość.
>sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAV
NNFEAHTINTVVHTNDSDKGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITID
SNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTAGQYQGLVSIILTKSTTTTTTTKGT
>sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVS
NTLVGVLTLSNTSIDTVSIASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDK
NAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGNYRANITITSTIKGGGTKKGTTDKKRozszerzenie fasta odnosi się do formatu pliku sekwencji. FASTA wywodzi się z oprogramowania bioinformatycznego FASTA i stąd wzięła swoją nazwę. Format FASTA ma wiele sekwencji ułożonych jedna po drugiej, a każda sekwencja będzie miała swój własny identyfikator, nazwę, opis i rzeczywiste dane sekwencji.
Step 2 - Utwórz nowy skrypt w języku Python, * simple_example.py ”, wprowadź poniższy kod i zapisz go.
from Bio.SeqIO import parse
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
file = open("example.fasta")
records = parse(file, "fasta") for record in records:
print("Id: %s" % record.id)
print("Name: %s" % record.name)
print("Description: %s" % record.description)
print("Annotations: %s" % record.annotations)
print("Sequence Data: %s" % record.seq)
print("Sequence Alphabet: %s" % record.seq.alphabet)Przyjrzyjmy się nieco dokładniej kodowi -
Line 1importuje klasę parsowania dostępną w module Bio.SeqIO. Moduł Bio.SeqIO służy do odczytu i zapisu pliku sekwencji w innym formacie, a klasa `parse 'służy do parsowania zawartości pliku sekwencji.
Line 2importuje klasę SeqRecord dostępną w module Bio.SeqRecord. Moduł ten służy do manipulowania rekordami sekwencji, a klasa SeqRecord jest używana do reprezentowania określonej sekwencji dostępnej w pliku sekwencji.
*Line 3"importuje klasę Seq dostępną w module Bio.Seq. Moduł ten służy do manipulowania danymi sekwencji, a klasa Seq jest używana do reprezentowania danych sekwencji określonego rekordu sekwencji dostępnego w pliku sekwencji.
Line 5 otwiera plik „example.fasta” używając zwykłej funkcji Pythona, otwórz.
Line 7 analizuje zawartość pliku sekwencji i zwraca zawartość jako listę obiektu SeqRecord.
Line 9-15 wykonuje pętlę nad rekordami za pomocą pętli Python for i drukuje atrybuty rekordu sekwencji (SqlRecord), takie jak identyfikator, nazwa, opis, dane sekwencji itp.
Line 15 wypisuje typ sekwencji przy użyciu klasy Alphabet.
Step 3 - Otwórz wiersz polecenia i przejdź do folderu zawierającego plik sekwencji „example.fasta” i uruchom poniższe polecenie -
> python simple_example.pyStep 4- Python uruchamia skrypt i wyświetla wszystkie dane sekwencji dostępne w przykładowym pliku „przyklad.fasta”. Dane wyjściowe będą podobne do następującej treści.
Id: sp|P25730|FMS1_ECOLI
Name: sp|P25730|FMS1_ECOLI
Decription: sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin)
Annotations: {}
Sequence Data: MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAVNNFEAHTINTVVHTNDSD
KGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITIDSNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTA
GQYQGLVSIILTKSTTTTTTTKGT
Sequence Alphabet: SingleLetterAlphabet()
Id: sp|P15488|FMS3_ECOLI
Name: sp|P15488|FMS3_ECOLI
Decription: sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin)
Annotations: {}
Sequence Data: MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVSNTLVGVLTLSNTSIDTVS
IASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDKNAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGN
YRANITITSTIKGGGTKKGTTDKK
Sequence Alphabet: SingleLetterAlphabet()W tym przykładzie widzieliśmy trzy klasy, parse, SeqRecord i Seq. Te trzy klasy zapewniają większość funkcji, o których dowiemy się w następnej sekcji.
Sekwencja to seria liter używanych do reprezentowania białka organizmu, DNA lub RNA. Jest reprezentowany przez klasę Seq. Klasa Seq jest zdefiniowana w module Bio.Seq.
Utwórzmy prostą sekwencję w Biopythonie, jak pokazano poniżej -
>>> from Bio.Seq import Seq
>>> seq = Seq("AGCT")
>>> seq
Seq('AGCT')
>>> print(seq)
AGCTTutaj stworzyliśmy prostą sekwencję białek AGCT a każda litera reprezentuje Alanine, Glicyna, Cysteine i Threonina.
Każdy obiekt Seq ma dwa ważne atrybuty -
data - rzeczywisty ciąg sekwencji (AGCT)
alfabet - używany do reprezentowania typu sekwencji. np. sekwencja DNA, sekwencja RNA itp. Domyślnie nie przedstawia żadnej sekwencji i ma charakter generyczny.
Moduł alfabetu
Obiekty Seq zawierają atrybut Alphabet, aby określić typ sekwencji, litery i możliwe operacje. Jest zdefiniowany w module Bio.Alphabet. Alfabet można zdefiniować jak poniżej -
>>> from Bio.Seq import Seq
>>> myseq = Seq("AGCT")
>>> myseq
Seq('AGCT')
>>> myseq.alphabet
Alphabet()Moduł Alphabet zawiera poniższe klasy reprezentujące różne typy sekwencji. Alphabet - klasa bazowa dla wszystkich typów alfabetów.
SingleLetterAlphabet - Ogólny alfabet z literami o rozmiarze jeden. Wywodzi się z Alphabet i wszystkie inne typy alfabetów wywodzą się z niego.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import single_letter_alphabet
>>> test_seq = Seq('AGTACACTGGT', single_letter_alphabet)
>>> test_seq
Seq('AGTACACTGGT', SingleLetterAlphabet())ProteinAlphabet - Ogólny jednoliterowy alfabet białkowy.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_protein
>>> test_seq = Seq('AGTACACTGGT', generic_protein)
>>> test_seq
Seq('AGTACACTGGT', ProteinAlphabet())NucleotideAlphabet - Ogólny jednoliterowy alfabet nukleotydów.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_nucleotide
>>> test_seq = Seq('AGTACACTGGT', generic_nucleotide) >>> test_seq
Seq('AGTACACTGGT', NucleotideAlphabet())DNAAlphabet - rodzajowy jednoliterowy alfabet DNA.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_dna
>>> test_seq = Seq('AGTACACTGGT', generic_dna)
>>> test_seq
Seq('AGTACACTGGT', DNAAlphabet())RNAAlphabet - Ogólny jednoliterowy alfabet RNA.
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import generic_rna
>>> test_seq = Seq('AGTACACTGGT', generic_rna)
>>> test_seq
Seq('AGTACACTGGT', RNAAlphabet())Moduł Biopython, Bio.Alphabet.IUPAC zapewnia podstawowe typy sekwencji zdefiniowane przez społeczność IUPAC. Zawiera następujące klasy -
IUPACProtein (protein) - Alfabet białek IUPAC składający się z 20 standardowych aminokwasów.
ExtendedIUPACProtein (extended_protein) - Rozszerzony jednoliterowy alfabet białka IUPAC, zawierający litery X.
IUPACAmbiguousDNA (ambiguous_dna) - Wieloznaczne DNA IUPAC.
IUPACUnambiguousDNA (unambiguous_dna) - Wielkie litery jednoznaczne DNA IUPAC (GATC).
ExtendedIUPACDNA (extended_dna) - Rozszerzony alfabet DNA IUPAC.
IUPACAmbiguousRNA (ambiguous_rna) - Wieloznaczne RNA IUPAC wielkimi literami.
IUPACUnambiguousRNA (unambiguous_rna) - Wielkie litery jednoznaczne RNA IUPAC (GAUC).
Rozważmy prosty przykład klasy IUPACProtein, jak pokazano poniżej -
>>> from Bio.Alphabet import IUPAC
>>> protein_seq = Seq("AGCT", IUPAC.protein)
>>> protein_seq
Seq('AGCT', IUPACProtein())
>>> protein_seq.alphabetPonadto Biopython udostępnia wszystkie dane konfiguracyjne związane z bioinformatyką za pośrednictwem modułu Bio.Data. Na przykład IUPACData.protein_letters zawiera możliwe litery alfabetu IUPACProtein.
>>> from Bio.Data import IUPACData
>>> IUPACData.protein_letters
'ACDEFGHIKLMNPQRSTVWY'Podstawowe operacje
Ta sekcja zawiera krótkie wyjaśnienie wszystkich podstawowych operacji dostępnych w klasie Seq. Sekwencje są podobne do ciągów znaków w Pythonie. Możemy wykonywać operacje na łańcuchach Pythona, takie jak krojenie, liczenie, konkatenacja, znajdowanie, dzielenie i rozbieranie w sekwencjach.
Użyj poniższych kodów, aby uzyskać różne wyniki.
To get the first value in sequence.
>>> seq_string = Seq("AGCTAGCT")
>>> seq_string[0]
'A'To print the first two values.
>>> seq_string[0:2]
Seq('AG')To print all the values.
>>> seq_string[ : ]
Seq('AGCTAGCT')To perform length and count operations.
>>> len(seq_string)
8
>>> seq_string.count('A')
2To add two sequences.
>>> from Bio.Alphabet import generic_dna, generic_protein
>>> seq1 = Seq("AGCT", generic_dna)
>>> seq2 = Seq("TCGA", generic_dna)
>>> seq1+seq2
Seq('AGCTTCGA', DNAAlphabet())Tutaj dwa powyższe obiekty sekwencji, seq1, seq2 są rodzajowymi sekwencjami DNA, więc możesz je dodać i stworzyć nową sekwencję. Nie możesz dodawać sekwencji z niekompatybilnymi alfabetami, takich jak sekwencja białka i sekwencja DNA, jak określono poniżej -
>>> dna_seq = Seq('AGTACACTGGT', generic_dna)
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> dna_seq + protein_seq
.....
.....
TypeError: Incompatible alphabets DNAAlphabet() and ProteinAlphabet()
>>>Aby dodać dwie lub więcej sekwencji, najpierw zapisz je na liście Pythona, a następnie pobierz za pomocą „pętli for” i na koniec dodaj razem, jak pokazano poniżej -
>>> from Bio.Alphabet import generic_dna
>>> list = [Seq("AGCT",generic_dna),Seq("TCGA",generic_dna),Seq("AAA",generic_dna)]
>>> for s in list:
... print(s)
...
AGCT
TCGA
AAA
>>> final_seq = Seq(" ",generic_dna)
>>> for s in list:
... final_seq = final_seq + s
...
>>> final_seq
Seq('AGCTTCGAAAA', DNAAlphabet())W poniższej sekcji podano różne kody, aby uzyskać dane wyjściowe w oparciu o wymagania.
To change the case of sequence.
>>> from Bio.Alphabet import generic_rna
>>> rna = Seq("agct", generic_rna)
>>> rna.upper()
Seq('AGCT', RNAAlphabet())To check python membership and identity operator.
>>> rna = Seq("agct", generic_rna)
>>> 'a' in rna
True
>>> 'A' in rna
False
>>> rna1 = Seq("AGCT", generic_dna)
>>> rna is rna1
FalseTo find single letter or sequence of letter inside the given sequence.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.find('G')
1
>>> protein_seq.find('GG')
8To perform splitting operation.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein)
>>> protein_seq.split('A')
[Seq('', ProteinAlphabet()), Seq('GU', ProteinAlphabet()),
Seq('C', ProteinAlphabet()), Seq('CUGGU', ProteinAlphabet())]To perform strip operations in the sequence.
>>> strip_seq = Seq(" AGCT ")
>>> strip_seq
Seq(' AGCT ')
>>> strip_seq.strip()
Seq('AGCT')W tym rozdziale omówimy niektóre z zaawansowanych funkcji sekwencji zapewnianych przez Biopython.
Uzupełnienie i dopełnienie odwrotne
Sekwencję nukleotydową można uzupełnić odwrotnie, aby uzyskać nową sekwencję. Ponadto, komplementowana sekwencja może być komplementowana odwrotnie, aby uzyskać oryginalną sekwencję. Biopython zapewnia dwie metody wykonania tej funkcji -complement i reverse_complement. Kod do tego jest podany poniżej -
>>> from Bio.Alphabet import IUPAC
>>> nucleotide = Seq('TCGAAGTCAGTC', IUPAC.ambiguous_dna)
>>> nucleotide.complement()
Seq('AGCTTCAGTCAG', IUPACAmbiguousDNA())
>>>W tym przypadku metoda complement () pozwala na uzupełnienie sekwencji DNA lub RNA. Metoda reverse_complement () uzupełnia i odwraca wynikową sekwencję od lewej do prawej. Jest to pokazane poniżej -
>>> nucleotide.reverse_complement()
Seq('GACTGACTTCGA', IUPACAmbiguousDNA())Biopython używa zmiennej ambiguous_dna_complement dostarczonej przez Bio.Data.IUPACData do wykonania operacji uzupełniania.
>>> from Bio.Data import IUPACData
>>> import pprint
>>> pprint.pprint(IUPACData.ambiguous_dna_complement) {
'A': 'T',
'B': 'V',
'C': 'G',
'D': 'H',
'G': 'C',
'H': 'D',
'K': 'M',
'M': 'K',
'N': 'N',
'R': 'Y',
'S': 'S',
'T': 'A',
'V': 'B',
'W': 'W',
'X': 'X',
'Y': 'R'}
>>>Zawartość GC
Przewiduje się, że skład zasad genomowego DNA (zawartość GC) znacząco wpłynie na funkcjonowanie genomu i ekologię gatunku. Zawartość GC to liczba nukleotydów GC podzielona przez całkowitą liczbę nukleotydów.
Aby uzyskać zawartość nukleotydów GC, zaimportuj następujący moduł i wykonaj następujące kroki -
>>> from Bio.SeqUtils import GC
>>> nucleotide = Seq("GACTGACTTCGA",IUPAC.unambiguous_dna)
>>> GC(nucleotide)
50.0Transkrypcja
Transkrypcja to proces zamiany sekwencji DNA na sekwencję RNA. Rzeczywisty proces transkrypcji biologicznej polega na wykonaniu odwrotnego dopełniacza (TCAG → CUGA) w celu uzyskania mRNA, traktując DNA jako nić matrycową. Jednak w bioinformatyce, a więc w Biopythonie, zazwyczaj pracujemy bezpośrednio z nicią kodującą i możemy uzyskać sekwencję mRNA, zmieniając literę T na U.
Prosty przykład powyższego jest następujący -
>>> from Bio.Seq import Seq
>>> from Bio.Seq import transcribe
>>> from Bio.Alphabet import IUPAC
>>> dna_seq = Seq("ATGCCGATCGTAT",IUPAC.unambiguous_dna) >>> transcribe(dna_seq)
Seq('AUGCCGAUCGUAU', IUPACUnambiguousRNA())
>>>Aby odwrócić transkrypcję, T zmienia się na U, jak pokazano na poniższym kodzie -
>>> rna_seq = transcribe(dna_seq)
>>> rna_seq.back_transcribe()
Seq('ATGCCGATCGTAT', IUPACUnambiguousDNA())Aby uzyskać nić matrycową DNA, reverse_complement the back transkrybowane RNA, jak podano poniżej -
>>> rna_seq.back_transcribe().reverse_complement()
Seq('ATACGATCGGCAT', IUPACUnambiguousDNA())Tłumaczenie
Translacja to proces translacji sekwencji RNA na sekwencję białka. Rozważ sekwencję RNA, jak pokazano poniżej -
>>> rna_seq = Seq("AUGGCCAUUGUAAU",IUPAC.unambiguous_rna)
>>> rna_seq
Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())Teraz zastosuj funkcję translate () do powyższego kodu -
>>> rna_seq.translate()
Seq('MAIV', IUPACProtein())Powyższa sekwencja RNA jest prosta. Rozważ sekwencję RNA, AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA i zastosuj translate () -
>>> rna = Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA', IUPAC.unambiguous_rna)
>>> rna.translate()
Seq('MAIVMGR*KGAR', HasStopCodon(IUPACProtein(), '*'))Tutaj kodony stop są oznaczone gwiazdką „*”.
W metodzie translate () możliwe jest zatrzymanie się na pierwszym kodonie stop. Aby to zrobić, możesz przypisać to_stop = True w translate () w następujący sposób -
>>> rna.translate(to_stop = True)
Seq('MAIVMGR', IUPACProtein())Tutaj kodon stop nie jest zawarty w wynikowej sekwencji, ponieważ go nie zawiera.
Tabela tłumaczeń
Strona Kody genetyczne NCBI zawiera pełną listę tabel tłumaczeń używanych przez Biopython. Zobaczmy przykład standardowej tabeli do wizualizacji kodu -
>>> from Bio.Data import CodonTable
>>> table = CodonTable.unambiguous_dna_by_name["Standard"]
>>> print(table)
Table 1 Standard, SGC0
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--
>>>Biopython używa tej tabeli do przetłumaczenia DNA na białko, a także do znalezienia kodonu Stop.
Biopython zapewnia moduł Bio.SeqIO do odczytu i zapisu sekwencji odpowiednio zi do pliku (dowolnego strumienia). Obsługuje prawie wszystkie formaty plików dostępne w bioinformatyce. Większość oprogramowania zapewnia różne podejście do różnych formatów plików. Jednak Biopython świadomie stosuje jedno podejście, aby przedstawić użytkownikowi przeanalizowane dane sekwencji za pośrednictwem obiektu SeqRecord.
Dowiedzmy się więcej o SeqRecord w następnej sekcji.
SeqRecord
Moduł Bio.SeqRecord zapewnia SeqRecord do przechowywania metainformacji o sekwencji, a także samych danych sekwencji, jak podano poniżej -
seq - To jest rzeczywista sekwencja.
id - Jest to podstawowy identyfikator danej sekwencji. Typ domyślny to ciąg.
name - jest to nazwa sekwencji. Typ domyślny to ciąg.
opis - Wyświetla czytelne dla człowieka informacje o sekwencji.
adnotacje - jest to słownik dodatkowych informacji o sekwencji.
SeqRecord można zaimportować w sposób określony poniżej
from Bio.SeqRecord import SeqRecordZrozummy niuanse analizy pliku sekwencji przy użyciu rzeczywistego pliku sekwencji w kolejnych sekcjach.
Parsowanie formatów plików sekwencji
W tej sekcji wyjaśniono, jak analizować dwa najpopularniejsze formaty plików sekwencji, FASTA i GenBank.
FASTA
FASTAto najbardziej podstawowy format pliku do przechowywania danych sekwencji. Pierwotnie FASTA jest pakietem oprogramowania do dopasowywania sekwencji DNA i białek opracowanym podczas wczesnej ewolucji bioinformatyki i używanym głównie do wyszukiwania podobieństwa sekwencji.
Biopython udostępnia przykładowy plik FASTA, do którego można uzyskać dostęp pod adresem https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Pobierz i zapisz ten plik w katalogu próbek Biopython jako ‘orchid.fasta’.
Moduł Bio.SeqIO zapewnia metodę parse () do przetwarzania plików sekwencji i może być importowany w następujący sposób -
from Bio.SeqIO import parseparse () zawiera dwa argumenty, pierwszy to uchwyt pliku, a drugi to format pliku.
>>> file = open('path/to/biopython/sample/orchid.fasta')
>>> for record in parse(file, "fasta"):
... print(record.id)
...
gi|2765658|emb|Z78533.1|CIZ78533
gi|2765657|emb|Z78532.1|CCZ78532
..........
..........
gi|2765565|emb|Z78440.1|PPZ78440
gi|2765564|emb|Z78439.1|PBZ78439
>>>Tutaj metoda parse () zwraca iterowalny obiekt, który zwraca SeqRecord przy każdej iteracji. Będąc iterowalnym, zapewnia wiele wyrafinowanych i łatwych metod oraz pozwala zobaczyć niektóre funkcje.
Kolejny()
metoda next () zwraca następny element dostępny w iterowalnym obiekcie, którego możemy użyć do uzyskania pierwszej sekwencji, jak podano poniżej -
>>> first_seq_record = next(SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta'))
>>> first_seq_record.id 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.name 'gi|2765658|emb|Z78533.1|CIZ78533'
>>> first_seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', SingleLetterAlphabet())
>>> first_seq_record.description 'gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> first_seq_record.annotations
{}
>>>Tutaj seq_record.annotations jest puste, ponieważ format FASTA nie obsługuje adnotacji sekwencji.
rozumienie listy
Możemy przekonwertować iterowalny obiekt na listę przy użyciu funkcji rozumienia list, jak podano poniżej
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> all_seq = [seq_record for seq_record in seq_iter] >>> len(all_seq)
94
>>>Tutaj użyliśmy metody len, aby uzyskać całkowitą liczbę. Możemy otrzymać sekwencję o maksymalnej długości w następujący sposób -
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> max_seq = max(len(seq_record.seq) for seq_record in seq_iter)
>>> max_seq
789
>>>Możemy również filtrować sekwencję za pomocą poniższego kodu -
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')
>>> seq_under_600 = [seq_record for seq_record in seq_iter if len(seq_record.seq) < 600]
>>> for seq in seq_under_600:
... print(seq.id)
...
gi|2765606|emb|Z78481.1|PIZ78481
gi|2765605|emb|Z78480.1|PGZ78480
gi|2765601|emb|Z78476.1|PGZ78476
gi|2765595|emb|Z78470.1|PPZ78470
gi|2765594|emb|Z78469.1|PHZ78469
gi|2765564|emb|Z78439.1|PBZ78439
>>>Zapisanie kolekcji obiektów SqlRecord (przeanalizowanych danych) do pliku jest tak proste, jak wywołanie metody SeqIO.write, jak poniżej -
file = open("converted.fasta", "w)
SeqIO.write(seq_record, file, "fasta")Tej metody można skutecznie użyć do konwersji formatu określonego poniżej -
file = open("converted.gbk", "w)
SeqIO.write(seq_record, file, "genbank")GenBank
Jest to bogatszy format sekwencji dla genów i zawiera pola dla różnego rodzaju adnotacji. Biopython udostępnia przykładowy plik GenBank, do którego można uzyskać dostęp pod adresemhttps://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Pobierz i zapisz plik w katalogu próbek Biopython jako ‘orchid.gbk’
Ponieważ Biopython zapewnia jedną funkcję, parsuj, aby przeanalizować cały format bioinformatyczny. Przetwarzanie formatu GenBank jest tak proste, jak zmiana opcji formatu w metodzie analizy.
Kod na to samo podano poniżej -
>>> from Bio import SeqIO
>>> from Bio.SeqIO import parse
>>> seq_record = next(parse(open('path/to/biopython/sample/orchid.gbk'),'genbank'))
>>> seq_record.id
'Z78533.1'
>>> seq_record.name
'Z78533'
>>> seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', IUPACAmbiguousDNA())
>>> seq_record.description
'C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA'
>>> seq_record.annotations {
'molecule_type': 'DNA',
'topology': 'linear',
'data_file_division': 'PLN',
'date': '30-NOV-2006',
'accessions': ['Z78533'],
'sequence_version': 1,
'gi': '2765658',
'keywords': ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'],
'source': 'Cypripedium irapeanum',
'organism': 'Cypripedium irapeanum',
'taxonomy': [
'Eukaryota',
'Viridiplantae',
'Streptophyta',
'Embryophyta',
'Tracheophyta',
'Spermatophyta',
'Magnoliophyta',
'Liliopsida',
'Asparagales',
'Orchidaceae',
'Cypripedioideae',
'Cypripedium'],
'references': [
Reference(title = 'Phylogenetics of the slipper orchids (Cypripedioideae:
Orchidaceae): nuclear rDNA ITS sequences', ...),
Reference(title = 'Direct Submission', ...)
]
}Sequence alignment jest procesem układania dwóch lub więcej sekwencji (sekwencji DNA, RNA lub białek) w określonej kolejności w celu określenia regionu podobieństwa między nimi.
Zidentyfikowanie podobnego regionu pozwala nam wnioskować o wielu informacjach, takich jak jakie cechy są zachowane między gatunkami, jak blisko są różne gatunki genetycznie, jak ewoluują gatunki itp. Biopython zapewnia obszerne wsparcie dla dopasowania sekwencji.
Poznajmy niektóre z ważnych funkcji udostępnionych przez Biopythona w tym rozdziale -
Parsing Sequence Alignment
Biopython dostarcza moduł Bio.AlignIO do odczytu i zapisu wyrównania sekwencji. W bioinformatyce dostępnych jest wiele formatów służących do określania danych dopasowania sekwencji, podobnych do wcześniej nauczonych danych o sekwencjach. Bio.AlignIO zapewnia API podobne do Bio.SeqIO, z wyjątkiem tego, że Bio.SeqIO działa na danych sekwencji, a Bio.AlignIO działa na danych dopasowania sekwencji.
Zanim zaczniemy się uczyć, pobierzmy przykładowy plik dopasowania sekwencji z Internetu.
Aby pobrać przykładowy plik, wykonaj poniższe czynności -
Step 1 - Otwórz swoją ulubioną przeglądarkę i przejdź do http://pfam.xfam.org/family/browsestronie internetowej. Wyświetli wszystkie rodziny Pfam w kolejności alfabetycznej.
Step 2- Wybierz jedną rodzinę z mniejszą liczbą wartości nasion. Zawiera minimalną ilość danych i umożliwia nam łatwą pracę z wyrównaniem. Tutaj wybraliśmy / kliknęliśmy PF18225 i otwiera się przejdź dohttp://pfam.xfam.org/family/PF18225 i pokazuje pełne szczegóły na ten temat, w tym dopasowanie sekwencji.
Step 3 - Przejdź do sekcji wyrównywania i pobierz plik wyrównywania sekwencji w formacie sztokholmskim (PF18225_seed.txt).
Spróbujmy odczytać pobrany plik wyrównania sekwencji za pomocą Bio.AlignIO jak poniżej -
Importuj moduł Bio.AlignIO
>>> from Bio import AlignIOCzytaj wyrównanie metodą odczytu. read służy do odczytu pojedynczych danych wyrównania dostępnych w danym pliku. Jeśli podany plik zawiera wiele wyrównania, możemy użyć metody parsowania. metoda parse zwraca iterowalny obiekt wyrównania podobny do metody parse w module Bio.SeqIO.
>>> alignment = AlignIO.read(open("PF18225_seed.txt"), "stockholm")Wydrukuj obiekt wyrównania.
>>> print(alignment)
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Możemy również sprawdzić sekwencje (SeqRecord) dostępne w dopasowaniu, a także poniżej -
>>> for align in alignment:
... print(align.seq)
...
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVATVANQLRGRKRRAFARHREGP
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADITA---RLDRRREHGEHGVRKKP
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMAPMLIALNYRNRESHAQVDKKP
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMAPLFKVLSFRNREDQGLVNNKP
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIMVLAPRLTAKHPYDKVQDRNRK
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVADLMRKLDLDRPFKKLERKNRT
>>>Wiele linii trasowania
Ogólnie większość plików dopasowywania sekwencji zawiera pojedyncze dane dopasowania i są one wystarczające do użycia readmetoda, aby go przeanalizować. W koncepcji dopasowania wielu sekwencji dwie lub więcej sekwencji jest porównywanych w celu uzyskania najlepszych dopasowań podciągów między nimi, co skutkuje dopasowaniem wielu sekwencji w jednym pliku.
Jeśli format dopasowania sekwencji wejściowej zawiera więcej niż jedno wyrównanie sekwencji, musimy użyć parse metoda zamiast read metoda określona poniżej -
>>> from Bio import AlignIO
>>> alignments = AlignIO.parse(open("PF18225_seed.txt"), "stockholm")
>>> print(alignments)
<generator object parse at 0x000001CD1C7E0360>
>>> for alignment in alignments:
... print(alignment)
...
SingleLetterAlphabet() alignment with 6 rows and 65 columns
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123
AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125
>>>Tutaj metoda parse zwraca iterowalny obiekt wyrównania i można ją iterować, aby uzyskać rzeczywiste wyrównania.
Wyrównanie sekwencji par
Pairwise sequence alignment porównuje tylko dwie sekwencje na raz i zapewnia najlepsze możliwe dopasowanie sekwencji. Pairwise jest łatwy do zrozumienia i wyjątkowy do wywnioskowania z wynikowego dopasowania sekwencji.
Biopython udostępnia specjalny moduł, Bio.pairwise2zidentyfikować sekwencję dopasowania metodą par. Biopython stosuje najlepszy algorytm do znalezienia sekwencji dopasowania i jest porównywalny z innym oprogramowaniem.
Napiszmy przykład, aby znaleźć dopasowanie sekwencji dwóch prostych i hipotetycznych sekwencji przy użyciu modułu par. Pomoże nam to zrozumieć koncepcję dopasowania sekwencji i jak ją zaprogramować za pomocą Biopythona.
Krok 1
Zaimportuj moduł pairwise2 poleceniem podanym poniżej -
>>> from Bio import pairwise2Krok 2
Utwórz dwie sekwencje, seq1 i seq2 -
>>> from Bio.Seq import Seq
>>> seq1 = Seq("ACCGGT")
>>> seq2 = Seq("ACGT")Krok 3
Wywołaj metodę pairwise2.align.globalxx wraz z sekwencjami seq1 i seq2, aby znaleźć wyrównania, używając poniższego wiersza kodu -
>>> alignments = pairwise2.align.globalxx(seq1, seq2)Tutaj, globalxxwykonuje właściwą pracę i znajduje wszystkie najlepsze możliwe dopasowania w podanych sekwencjach. W rzeczywistości Bio.pairwise2 zapewnia całkiem zestaw metod, które są zgodne z poniższą konwencją, aby znaleźć wyrównania w różnych scenariuszach.
<sequence alignment type>XYTutaj typ przyrównania sekwencji odnosi się do typu przyrównania, który może być globalny lub lokalny. typ globalny polega na znajdowaniu dopasowania sekwencji, biorąc pod uwagę całą sekwencję. typ lokalny polega na znalezieniu dopasowania sekwencji poprzez sprawdzenie również podzbioru danych sekwencji. Będzie to żmudne, ale pozwoli lepiej zrozumieć podobieństwo między podanymi sekwencjami.
X odnosi się do wyniku dopasowania. Możliwe wartości to x (dopasowanie ścisłe), m (wynik oparty na identycznych znakach), d (słownik dostarczony przez użytkownika ze znakiem i wynikiem dopasowania) i wreszcie c (funkcja zdefiniowana przez użytkownika zapewniająca niestandardowy algorytm oceniania).
Y odnosi się do kary za przerwę. Możliwe wartości to x (brak kar za przerwy), s (te same kary dla obu sekwencji), d (różne kary za każdą sekwencję) i wreszcie c (funkcja zdefiniowana przez użytkownika w celu zapewnienia niestandardowych kar za przerwy)
Tak więc, localds jest również prawidłową metodą, która znajduje dopasowanie sekwencji przy użyciu techniki lokalnego dopasowania, słownika dostarczonego przez użytkownika dla dopasowań i kary za przerwy zapewniane przez użytkownika dla obu sekwencji.
>>> test_alignments = pairwise2.align.localds(seq1, seq2, blosum62, -10, -1)Tutaj blosum62 odnosi się do słownika dostępnego w module pairwise2 w celu zapewnienia wyniku dopasowania. -10 odnosi się do kary za przerwę, a -1 do kary za wydłużenie przerwy.
Krok 4
Zrób pętlę nad iterowalnym obiektem wyrównywania, pobierz każdy indywidualny obiekt wyrównania i wydrukuj go.
>>> for alignment in alignments:
... print(alignment)
...
('ACCGGT', 'A-C-GT', 4.0, 0, 6)
('ACCGGT', 'AC--GT', 4.0, 0, 6)
('ACCGGT', 'A-CG-T', 4.0, 0, 6)
('ACCGGT', 'AC-G-T', 4.0, 0, 6)Krok 5
Moduł Bio.pairwise2 zapewnia metodę formatowania, format_alignment w celu lepszej wizualizacji wyniku -
>>> from Bio.pairwise2 import format_alignment
>>> alignments = pairwise2.align.globalxx(seq1, seq2)
>>> for alignment in alignments:
... print(format_alignment(*alignment))
...
ACCGGT
| | ||
A-C-GT
Score=4
ACCGGT
|| ||
AC--GT
Score=4
ACCGGT
| || |
A-CG-T
Score=4
ACCGGT
|| | |
AC-G-T
Score=4
>>>Biopython dostarcza również inny moduł do wyrównywania sekwencji, Align. Ten moduł zapewnia inny zestaw API do prostego ustawiania parametrów, takich jak algorytm, tryb, wynik dopasowania, kary za przerwy itp. Prosty wygląd obiektu Align jest następujący:
>>> from Bio import Align
>>> aligner = Align.PairwiseAligner()
>>> print(aligner)
Pairwise sequence aligner with parameters
match score: 1.000000
mismatch score: 0.000000
target open gap score: 0.000000
target extend gap score: 0.000000
target left open gap score: 0.000000
target left extend gap score: 0.000000
target right open gap score: 0.000000
target right extend gap score: 0.000000
query open gap score: 0.000000
query extend gap score: 0.000000
query left open gap score: 0.000000
query left extend gap score: 0.000000
query right open gap score: 0.000000
query right extend gap score: 0.000000
mode: global
>>>Obsługa narzędzi do wyrównywania sekwencji
Biopython zapewnia interfejs do wielu narzędzi do dopasowywania sekwencji poprzez moduł Bio.Align.Applications. Niektóre z narzędzi są wymienione poniżej -
- ClustalW
- MUSCLE
- Igła EMBOSS i woda
Napiszmy prosty przykład w Biopythonie, aby stworzyć wyrównanie sekwencji za pomocą najpopularniejszego narzędzia do dopasowywania, ClustalW.
Step 1 - Pobierz program Clustalw z witryny http://www.clustal.org/download/current/i zainstaluj. Zaktualizuj również ścieżkę systemową PATH, używając „klastrowej” ścieżki instalacji.
Step 2 - importuj ClustalwCommanLine z modułu Bio.Align.Applications.
>>> from Bio.Align.Applications import ClustalwCommandlineStep 3 - Ustaw cmd, wywołując ClustalwCommanLine z plikiem wejściowym opuntia.fasta dostępnym w pakiecie Biopython. https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/opuntia.fasta
>>> cmd = ClustalwCommandline("clustalw2",
infile="/path/to/biopython/sample/opuntia.fasta")
>>> print(cmd)
clustalw2 -infile=fasta/opuntia.fastaStep 4 - Wywołanie cmd () uruchomi polecenie clustalw i wyświetli wynikowy plik wyrównania, opuntia.aln.
>>> stdout, stderr = cmd()Step 5 - Przeczytaj i wydrukuj plik wyrównania, jak poniżej -
>>> from Bio import AlignIO
>>> align = AlignIO.read("/path/to/biopython/sample/opuntia.aln", "clustal")
>>> print(align)
SingleLetterAlphabet() alignment with 7 rows and 906 columns
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273285|gb|AF191659.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273284|gb|AF191658.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273287|gb|AF191661.1|AF191
TATACATAAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273286|gb|AF191660.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273290|gb|AF191664.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273289|gb|AF191663.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273291|gb|AF191665.1|AF191
>>>BLAST oznacza Basic Local Alignment Search Tool. Znajduje regiony podobieństwa między sekwencjami biologicznymi. Biopython dostarcza moduł Bio.Blast do obsługi operacji NCBI BLAST. Możesz uruchomić BLAST w połączeniu lokalnym lub przez połączenie internetowe.
Zrozummy w skrócie te dwa połączenia w następnej sekcji -
Bieganie przez Internet
Biopython dostarcza moduł Bio.Blast.NCBIWWW do wywoływania wersji online BLAST. Aby to zrobić, musimy zaimportować następujący moduł -
>>> from Bio.Blast import NCBIWWWModuł NCBIWW zapewnia funkcję qblast do sprawdzania wersji online BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi. qblast obsługuje wszystkie parametry obsługiwane przez wersję online.
Aby uzyskać pomoc dotyczącą tego modułu, użyj poniższego polecenia i zapoznaj się z funkcjami -
>>> help(NCBIWWW.qblast)
Help on function qblast in module Bio.Blast.NCBIWWW:
qblast(
program, database, sequence,
url_base = 'https://blast.ncbi.nlm.nih.gov/Blast.cgi',
auto_format = None,
composition_based_statistics = None,
db_genetic_code = None,
endpoints = None,
entrez_query = '(none)',
expect = 10.0,
filter = None,
gapcosts = None,
genetic_code = None,
hitlist_size = 50,
i_thresh = None,
layout = None,
lcase_mask = None,
matrix_name = None,
nucl_penalty = None,
nucl_reward = None,
other_advanced = None,
perc_ident = None,
phi_pattern = None,
query_file = None,
query_believe_defline = None,
query_from = None,
query_to = None,
searchsp_eff = None,
service = None,
threshold = None,
ungapped_alignment = None,
word_size = None,
alignments = 500,
alignment_view = None,
descriptions = 500,
entrez_links_new_window = None,
expect_low = None,
expect_high = None,
format_entrez_query = None,
format_object = None,
format_type = 'XML',
ncbi_gi = None,
results_file = None,
show_overview = None,
megablast = None,
template_type = None,
template_length = None
)
BLAST search using NCBI's QBLAST server or a cloud service provider.
Supports all parameters of the qblast API for Put and Get.
Please note that BLAST on the cloud supports the NCBI-BLAST Common
URL API (http://ncbi.github.io/blast-cloud/dev/api.html).
To use this feature, please set url_base to 'http://host.my.cloud.service.provider.com/cgi-bin/blast.cgi' and
format_object = 'Alignment'. For more details, please see 8. Biopython – Overview of BLAST
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE = BlastDocs&DOC_TYPE = CloudBlast
Some useful parameters:
- program blastn, blastp, blastx, tblastn, or tblastx (lower case)
- database Which database to search against (e.g. "nr").
- sequence The sequence to search.
- ncbi_gi TRUE/FALSE whether to give 'gi' identifier.
- descriptions Number of descriptions to show. Def 500.
- alignments Number of alignments to show. Def 500.
- expect An expect value cutoff. Def 10.0.
- matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45).
- filter "none" turns off filtering. Default no filtering
- format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML".
- entrez_query Entrez query to limit Blast search
- hitlist_size Number of hits to return. Default 50
- megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only)
- service plain, psi, phi, rpsblast, megablast (lower case)
This function does no checking of the validity of the parameters
and passes the values to the server as is. More help is available at:
https://ncbi.github.io/blast-cloud/dev/api.htmlZwykle argumenty funkcji qblast są w zasadzie analogiczne do różnych parametrów, które można ustawić na stronie WWW BLAST. To sprawia, że funkcja qblast jest łatwa do zrozumienia, a także zmniejsza krzywą uczenia się, aby z niej korzystać.
Łączenie i wyszukiwanie
Aby zrozumieć proces łączenia i wyszukiwania wersji online BLAST, przeprowadźmy proste wyszukiwanie sekwencji (dostępne w naszym lokalnym pliku sekwencji) na serwerze online BLAST poprzez Biopython.
Step 1 - Utwórz plik o nazwie blast_example.fasta w katalogu Biopython i podaj poniższe informacje o sekwencji jako dane wejściowe
Example of a single sequence in FASTA/Pearson format:
>sequence A ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattcatat
tctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtc
>sequence B ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattca
tattctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtcStep 2 - Importuj moduł NCBIWWW.
>>> from Bio.Blast import NCBIWWWStep 3 - Otwórz plik sekwencji, blast_example.fasta przy użyciu modułu IO Pythona.
>>> sequence_data = open("blast_example.fasta").read()
>>> sequence_data
'Example of a single sequence in FASTA/Pearson format:\n\n\n> sequence
A\nggtaagtcctctagtacaaacacccccaatattgtgatataattaaaatt
atattcatat\ntctgttgccagaaaaaacacttttaggctatattagagccatcttctttg aagcgttgtc\n\n'Step 4- Teraz wywołaj funkcję qblast przekazującą dane sekwencji jako główny parametr. Drugi parametr reprezentuje bazę danych (nt) i program wewnętrzny (blastn).
>>> result_handle = NCBIWWW.qblast("blastn", "nt", sequence_data)
>>> result_handle
<_io.StringIO object at 0x000001EC9FAA4558>blast_resultszawiera wynik naszego wyszukiwania. Można go zapisać w pliku do późniejszego wykorzystania, a także przeanalizować, aby uzyskać szczegółowe informacje. Dowiemy się, jak to zrobić w następnej sekcji.
Step 5 - Tę samą funkcjonalność można wykonać za pomocą obiektu Seq, zamiast używać całego pliku fasta, jak pokazano poniżej -
>>> from Bio import SeqIO
>>> seq_record = next(SeqIO.parse(open('blast_example.fasta'),'fasta'))
>>> seq_record.id
'sequence'
>>> seq_record.seq
Seq('ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatat...gtc',
SingleLetterAlphabet())Teraz wywołaj funkcję qblast przekazując obiekt Seq, record.seq jako główny parametr.
>>> result_handle = NCBIWWW.qblast("blastn", "nt", seq_record.seq)
>>> print(result_handle)
<_io.StringIO object at 0x000001EC9FAA4558>BLAST automatycznie przypisze identyfikator do twojej sekwencji.
Step 6 - obiekt result_handle będzie miał cały wynik i można go zapisać do pliku do późniejszego wykorzystania.
>>> with open('results.xml', 'w') as save_file:
>>> blast_results = result_handle.read()
>>> save_file.write(blast_results)Zobaczymy, jak przeanalizować plik wynikowy w dalszej części.
Uruchamianie samodzielnego BLASTa
Ta sekcja wyjaśnia jak uruchomić BLAST w systemie lokalnym. Jeśli uruchomisz BLAST w systemie lokalnym, może być szybszy, a także pozwala na stworzenie własnej bazy danych do przeszukiwania sekwencji.
Łączenie BLAST
Ogólnie, lokalne uruchamianie BLAST-a nie jest zalecane ze względu na jego duży rozmiar, dodatkowy wysiłek potrzebny do uruchomienia oprogramowania i związane z tym koszty. Online BLAST jest wystarczające do podstawowych i zaawansowanych celów. Oczywiście czasami może być konieczne zainstalowanie go lokalnie.
Weź pod uwagę, że często wyszukujesz w Internecie, co może wymagać dużo czasu i dużego obciążenia sieci, a jeśli masz zastrzeżone dane sekwencyjne lub problemy związane z IP, zaleca się zainstalowanie go lokalnie.
Aby to zrobić, musimy wykonać poniższe kroki -
Step 1- Pobierz i zainstaluj najnowszy plik binarny blast za pomocą podanego linku - ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Step 2- Pobierz i rozpakuj najnowszą i niezbędną bazę danych, korzystając z poniższego linku - ftp://ftp.ncbi.nlm.nih.gov/blast/db/
Oprogramowanie BLAST udostępnia wiele baz danych w swojej witrynie. Miejmy pobrać alu.n.gz plik ze strony bazy podmuch i rozpakować go do folderu alu. Ten plik jest w formacie FASTA. Aby użyć tego pliku w naszej aplikacji do obróbki strumieniowej, musimy najpierw przekonwertować plik z formatu FASTA na format bazy danych do obsługi strumieniowej. BLAST zapewnia aplikację makeblastdb do wykonania tej konwersji.
Użyj poniższego fragmentu kodu -
cd /path/to/alu
makeblastdb -in alu.n -parse_seqids -dbtype nucl -out alunUruchomienie powyższego kodu przeanalizuje plik wejściowy alu.n i utworzy bazę danych BLAST jako wiele plików alun.nsq, alun.nsi itd. Teraz możemy przeszukać tę bazę danych, aby znaleźć sekwencję.
Zainstalowaliśmy BLAST na naszym lokalnym serwerze, a także mamy przykładową bazę danych BLAST, alun zapytać przeciwko temu.
Step 3- Utwórzmy przykładowy plik sekwencji, aby wysłać zapytanie do bazy danych. Utwórz plik search.fsa i umieść w nim poniższe dane.
>gnl|alu|Z15030_HSAL001056 (Alu-J)
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCT
TGAGCCTAGGAGTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAA
AGAAAAAAAAAATAGCTCTGCTGGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTG
GGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCCACGATCACACCACT
GCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
>gnl|alu|D00596_HSAL003180 (Alu-Sx)
AGCCAGGTGTGGTGGCTCACGCCTGTAATCCCACCGCTTTGGGAGGCTGAGTCAGATCAC
CTGAGGTTAGGAATTTGGGACCAGCCTGGCCAACATGGCGACACCCCAGTCTCTACTAAT
AACACAAAAAATTAGCCAGGTGTGCTGGTGCATGTCTGTAATCCCAGCTACTCAGGAGGC
TGAGGCATGAGAATTGCTCACGAGGCGGAGGTTGTAGTGAGCTGAGATCGTGGCACTGTA
CTCCAGCCTGGCGACAGAGGGAGAACCCATGTCAAAAACAAAAAAAGACACCACCAAAGG
TCAAAGCATA
>gnl|alu|X55502_HSAL000745 (Alu-J)
TGCCTTCCCCATCTGTAATTCTGGCACTTGGGGAGTCCAAGGCAGGATGATCACTTATGC
CCAAGGAATTTGAGTACCAAGCCTGGGCAATATAACAAGGCCCTGTTTCTACAAAAACTT
TAAACAATTAGCCAGGTGTGGTGGTGCGTGCCTGTGTCCAGCTACTCAGGAAGCTGAGGC
AAGAGCTTGAGGCTACAGTGAGCTGTGTTCCACCATGGTGCTCCAGCCTGGGTGACAGGG
CAAGACCCTGTCAAAAGAAAGGAAGAAAGAACGGAAGGAAAGAAGGAAAGAAACAAGGAG
AGDane o sekwencji są zbierane z pliku alu.n; dlatego pasuje do naszej bazy danych.
Step 4 - Oprogramowanie BLAST udostępnia wiele aplikacji do przeszukiwania bazy danych i używamy blastn. blastn application requires minimum of three arguments, db, query and out. db odwołuje się do bazy danych przeciwko wyszukiwaniu; query to sekwencja do dopasowania i outto plik do przechowywania wyników. Teraz uruchom poniższe polecenie, aby wykonać to proste zapytanie -
blastn -db alun -query search.fsa -out results.xml -outfmt 5Uruchomienie powyższego polecenia spowoduje wyszukanie i wyświetlenie danych wyjściowych w results.xml plik jak podano poniżej (częściowo dane) -
<?xml version = "1.0"?>
<!DOCTYPE BlastOutput PUBLIC "-//NCBI//NCBI BlastOutput/EN"
"http://www.ncbi.nlm.nih.gov/dtd/NCBI_BlastOutput.dtd">
<BlastOutput>
<BlastOutput_program>blastn</BlastOutput_program>
<BlastOutput_version>BLASTN 2.7.1+</BlastOutput_version>
<BlastOutput_reference>Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
</BlastOutput_reference>
<BlastOutput_db>alun</BlastOutput_db>
<BlastOutput_query-ID>Query_1</BlastOutput_query-ID>
<BlastOutput_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</BlastOutput_query-def>
<BlastOutput_query-len>292</BlastOutput_query-len>
<BlastOutput_param>
<Parameters>
<Parameters_expect>10</Parameters_expect>
<Parameters_sc-match>1</Parameters_sc-match>
<Parameters_sc-mismatch>-2</Parameters_sc-mismatch>
<Parameters_gap-open>0</Parameters_gap-open>
<Parameters_gap-extend>0</Parameters_gap-extend>
<Parameters_filter>L;m;</Parameters_filter>
</Parameters>
</BlastOutput_param>
<BlastOutput_iterations>
<Iteration>
<Iteration_iter-num>1</Iteration_iter-num><Iteration_query-ID>Query_1</Iteration_query-ID>
<Iteration_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</Iteration_query-def>
<Iteration_query-len>292</Iteration_query-len>
<Iteration_hits>
<Hit>
<Hit_num>1</Hit_num>
<Hit_id>gnl|alu|Z15030_HSAL001056</Hit_id>
<Hit_def>(Alu-J)</Hit_def>
<Hit_accession>Z15030_HSAL001056</Hit_accession>
<Hit_len>292</Hit_len>
<Hit_hsps>
<Hsp>
<Hsp_num>1</Hsp_num>
<Hsp_bit-score>540.342</Hsp_bit-score>
<Hsp_score>292</Hsp_score>
<Hsp_evalue>4.55414e-156</Hsp_evalue>
<Hsp_query-from>1</Hsp_query-from>
<Hsp_query-to>292</Hsp_query-to>
<Hsp_hit-from>1</Hsp_hit-from>
<Hsp_hit-to>292</Hsp_hit-to>
<Hsp_query-frame>1</Hsp_query-frame>
<Hsp_hit-frame>1</Hsp_hit-frame>
<Hsp_identity>292</Hsp_identity>
<Hsp_positive>292</Hsp_positive>
<Hsp_gaps>0</Hsp_gaps>
<Hsp_align-len>292</Hsp_align-len>
<Hsp_qseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGAGTTTG
CGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCTGGTGGTGCATG
CCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCC
ACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
</Hsp_qseq>
<Hsp_hseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGA
GTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCT
GGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGG
CTGTGGTGAGCCACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAAC
AAATAA
</Hsp_hseq>
<Hsp_midline>
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||
</Hsp_midline>
</Hsp>
</Hit_hsps>
</Hit>
.........................
.........................
.........................
</Iteration_hits>
<Iteration_stat>
<Statistics>
<Statistics_db-num>327</Statistics_db-num>
<Statistics_db-len>80506</Statistics_db-len>
<Statistics_hsp-lenv16</Statistics_hsp-len>
<Statistics_eff-space>21528364</Statistics_eff-space>
<Statistics_kappa>0.46</Statistics_kappa>
<Statistics_lambda>1.28</Statistics_lambda>
<Statistics_entropy>0.85</Statistics_entropy>
</Statistics>
</Iteration_stat>
</Iteration>
</BlastOutput_iterations>
</BlastOutput>Powyższe polecenie można uruchomić w Pythonie za pomocą poniższego kodu -
>>> from Bio.Blast.Applications import NcbiblastnCommandline
>>> blastn_cline = NcbiblastnCommandline(query = "search.fasta", db = "alun",
outfmt = 5, out = "results.xml")
>>> stdout, stderr = blastn_cline()Tutaj pierwszy z nich jest uchwytem do wyjścia wybuchu, a drugi jest możliwym wyjściem błędu wygenerowanym przez polecenie wybuchu.
Ponieważ udostępniliśmy plik wyjściowy jako argument wiersza poleceń (out = „results.xml”) i ustawiliśmy format wyjściowy na XML (outfmt = 5), plik wyjściowy zostanie zapisany w bieżącym katalogu roboczym.
Analiza wyniku BLAST
Generalnie dane wyjściowe BLAST są analizowane jako format XML przy użyciu modułu NCBIXML. Aby to zrobić, musimy zaimportować następujący moduł -
>>> from Bio.Blast import NCBIXMLTeraz, open the file directly using python open method i use NCBIXML parse method jak podano poniżej -
>>> E_VALUE_THRESH = 1e-20
>>> for record in NCBIXML.parse(open("results.xml")):
>>> if record.alignments:
>>> print("\n")
>>> print("query: %s" % record.query[:100])
>>> for align in record.alignments:
>>> for hsp in align.hsps:
>>> if hsp.expect < E_VALUE_THRESH:
>>> print("match: %s " % align.title[:100])To da następujący wynik:
query: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|Z15030_HSAL001056 (Alu-J)
match: gnl|alu|L12964_HSAL003860 (Alu-J)
match: gnl|alu|L13042_HSAL003863 (Alu-FLA?)
match: gnl|alu|M86249_HSAL001462 (Alu-FLA?)
match: gnl|alu|M29484_HSAL002265 (Alu-J)
query: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|D00596_HSAL003180 (Alu-Sx)
match: gnl|alu|J03071_HSAL001860 (Alu-J)
match: gnl|alu|X72409_HSAL005025 (Alu-Sx)
query: gnl|alu|X55502_HSAL000745 (Alu-J)
match: gnl|alu|X55502_HSAL000745 (Alu-J)Entrezto internetowy system wyszukiwania udostępniany przez NCBI. Zapewnia dostęp do prawie wszystkich znanych baz danych biologii molekularnej ze zintegrowanym globalnym zapytaniem obsługującym operatory boolowskie i wyszukiwanie w terenie. Zwraca wyniki ze wszystkich baz danych z informacjami, takimi jak liczba trafień z każdej bazy danych, rekordy z linkami do źródłowej bazy danych itp.
Niektóre z popularnych baz danych, do których można uzyskać dostęp przez Entrez, są wymienione poniżej -
- Pubmed
- Pubmed Central
- Nukleotyd (baza danych sekwencji GenBank)
- Białko (baza danych sekwencji)
- Genom (baza danych całego genomu)
- Struktura (trójwymiarowa struktura makromolekularna)
- Taksonomia (organizmy w GenBank)
- SNP (polimorfizm pojedynczego nukleotydu)
- UniGene (klastry sekwencji transkrypcyjnych zorientowanych genowo)
- CDD (baza danych konserwowanych domen białkowych)
- Domeny 3D (domeny ze struktury Entrez)
Oprócz powyższych baz danych, Entrez zapewnia o wiele więcej baz danych do wyszukiwania pól.
Biopython zapewnia specjalny moduł Entrez, Bio.Entrez, aby uzyskać dostęp do bazy danych Entrez. Nauczmy się, jak uzyskać dostęp do Entrez za pomocą Biopythona w tym rozdziale -
Kroki połączenia z bazą danych
Aby dodać funkcje Entrez, zaimportuj następujący moduł -
>>> from Bio import EntrezNastępnie ustaw swój adres e-mail, aby zidentyfikować, kto jest powiązany z kodem podanym poniżej -
>>> Entrez.email = '<youremail>'Następnie ustaw parametr narzędzia Entrez i domyślnie jest to Biopython.
>>> Entrez.tool = 'Demoscript'Teraz, call einfo function to find index term counts, last update, and available links for each database jak określono poniżej -
>>> info = Entrez.einfo()Metoda einfo zwraca obiekt, który zapewnia dostęp do informacji poprzez metodę odczytu, jak pokazano poniżej -
>>> data = info.read()
>>> print(data)
<?xml version = "1.0" encoding = "UTF-8" ?>
<!DOCTYPE eInfoResult PUBLIC "-//NLM//DTD einfo 20130322//EN"
"https://eutils.ncbi.nlm.nih.gov/eutils/dtd/20130322/einfo.dtd">
<eInfoResult>
<DbList>
<DbName>pubmed</DbName>
<DbName>protein</DbName>
<DbName>nuccore</DbName>
<DbName>ipg</DbName>
<DbName>nucleotide</DbName>
<DbName>nucgss</DbName>
<DbName>nucest</DbName>
<DbName>structure</DbName>
<DbName>sparcle</DbName>
<DbName>genome</DbName>
<DbName>annotinfo</DbName>
<DbName>assembly</DbName>
<DbName>bioproject</DbName>
<DbName>biosample</DbName>
<DbName>blastdbinfo</DbName>
<DbName>books</DbName>
<DbName>cdd</DbName>
<DbName>clinvar</DbName>
<DbName>clone</DbName>
<DbName>gap</DbName>
<DbName>gapplus</DbName>
<DbName>grasp</DbName>
<DbName>dbvar</DbName>
<DbName>gene</DbName>
<DbName>gds</DbName>
<DbName>geoprofiles</DbName>
<DbName>homologene</DbName>
<DbName>medgen</DbName>
<DbName>mesh</DbName>
<DbName>ncbisearch</DbName>
<DbName>nlmcatalog</DbName>
<DbName>omim</DbName>
<DbName>orgtrack</DbName>
<DbName>pmc</DbName>
<DbName>popset</DbName>
<DbName>probe</DbName>
<DbName>proteinclusters</DbName>
<DbName>pcassay</DbName>
<DbName>biosystems</DbName>
<DbName>pccompound</DbName>
<DbName>pcsubstance</DbName>
<DbName>pubmedhealth</DbName>
<DbName>seqannot</DbName>
<DbName>snp</DbName>
<DbName>sra</DbName>
<DbName>taxonomy</DbName>
<DbName>biocollections</DbName>
<DbName>unigene</DbName>
<DbName>gencoll</DbName>
<DbName>gtr</DbName>
</DbList>
</eInfoResult>Dane są w formacie XML i aby pobrać dane jako obiekt Pythona, użyj Entrez.read metody tak szybko, jak Entrez.einfo() wywoływana jest metoda -
>>> info = Entrez.einfo()
>>> record = Entrez.read(info)Tutaj rekord jest słownikiem, który ma jeden klucz, DbList, jak pokazano poniżej -
>>> record.keys()
[u'DbList']Dostęp do klawisza DbList zwraca listę nazw baz danych pokazaną poniżej -
>>> record[u'DbList']
['pubmed', 'protein', 'nuccore', 'ipg', 'nucleotide', 'nucgss',
'nucest', 'structure', 'sparcle', 'genome', 'annotinfo', 'assembly',
'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar',
'clone', 'gap', 'gapplus', 'grasp', 'dbvar', 'gene', 'gds', 'geoprofiles',
'homologene', 'medgen', 'mesh', 'ncbisearch', 'nlmcatalog', 'omim',
'orgtrack', 'pmc', 'popset', 'probe', 'proteinclusters', 'pcassay',
'biosystems', 'pccompound', 'pcsubstance', 'pubmedhealth', 'seqannot',
'snp', 'sra', 'taxonomy', 'biocollections', 'unigene', 'gencoll', 'gtr']
>>>Zasadniczo moduł Entrez analizuje XML zwracany przez system wyszukiwania Entrez i dostarcza go jako słownik i listy Pythona.
Przeszukaj bazę danych
Aby przeszukać jedną z baz danych Entrez, możemy użyć modułu Bio.Entrez.esearch (). Jest zdefiniowany poniżej -
>>> info = Entrez.einfo()
>>> info = Entrez.esearch(db = "pubmed",term = "genome")
>>> record = Entrez.read(info)
>>>print(record)
DictElement({u'Count': '1146113', u'RetMax': '20', u'IdList':
['30347444', '30347404', '30347317', '30347292',
'30347286', '30347249', '30347194', '30347187',
'30347172', '30347088', '30347075', '30346992',
'30346990', '30346982', '30346980', '30346969',
'30346962', '30346954', '30346941', '30346939'],
u'TranslationStack': [DictElement({u'Count':
'927819', u'Field': 'MeSH Terms', u'Term': '"genome"[MeSH Terms]',
u'Explode': 'Y'}, attributes = {})
, DictElement({u'Count': '422712', u'Field':
'All Fields', u'Term': '"genome"[All Fields]', u'Explode': 'N'}, attributes = {}),
'OR', 'GROUP'], u'TranslationSet': [DictElement({u'To': '"genome"[MeSH Terms]
OR "genome"[All Fields]', u'From': 'genome'}, attributes = {})], u'RetStart': '0',
u'QueryTranslation': '"genome"[MeSH Terms] OR "genome"[All Fields]'},
attributes = {})
>>>Jeśli przypiszesz nieprawidłową bazę danych, to zwraca
>>> info = Entrez.esearch(db = "blastdbinfo",term = "books")
>>> record = Entrez.read(info)
>>> print(record)
DictElement({u'Count': '0', u'RetMax': '0', u'IdList': [],
u'WarningList': DictElement({u'OutputMessage': ['No items found.'],
u'PhraseIgnored': [], u'QuotedPhraseNotFound': []}, attributes = {}),
u'ErrorList': DictElement({u'FieldNotFound': [], u'PhraseNotFound':
['books']}, attributes = {}), u'TranslationSet': [], u'RetStart': '0',
u'QueryTranslation': '(books[All Fields])'}, attributes = {})Jeśli chcesz przeszukiwać bazę danych, możesz użyć Entrez.egquery. To jest podobne doEntrez.esearch poza tym, że wystarczy podać słowo kluczowe i pominąć parametr bazy danych.
>>>info = Entrez.egquery(term = "entrez")
>>> record = Entrez.read(info)
>>> for row in record["eGQueryResult"]:
... print(row["DbName"], row["Count"])
...
pubmed 458
pmc 12779 mesh 1
...
...
...
biosample 7
biocollections 0Pobierz rekordy
Enterz zapewnia specjalną metodę, efetch do wyszukiwania i pobierania pełnych szczegółów rekordu z Entrez. Rozważmy następujący prosty przykład -
>>> handle = Entrez.efetch(
db = "nucleotide", id = "EU490707", rettype = "fasta")Teraz możemy po prostu odczytać rekordy za pomocą obiektu SeqIO
>>> record = SeqIO.read( handle, "fasta" )
>>> record
SeqRecord(seq = Seq('ATTTTTTACGAACCTGTGGAAATTTTTGGTTATGACAATAAATCTAGTTTAGTA...GAA',
SingleLetterAlphabet()), id = 'EU490707.1', name = 'EU490707.1',
description = 'EU490707.1
Selenipedium aequinoctiale maturase K (matK) gene, partial cds; chloroplast',
dbxrefs = [])Biopython dostarcza moduł Bio.PDB do manipulacji strukturami polipeptydowymi. PDB (Protein Data Bank) jest największym źródłem informacji o strukturze białek dostępnym online. Zawiera wiele różnych struktur białkowych, w tym kompleksy białko-białko, białko-DNA, białko-RNA.
Aby załadować PDB, wpisz poniższe polecenie -
from Bio.PDB import *Formaty plików struktury białek
PDB rozprowadza struktury białkowe w trzech różnych formatach -
- Format pliku oparty na XML, który nie jest obsługiwany przez Biopython
- Format pliku pdb, który jest specjalnie sformatowanym plikiem tekstowym
- Format plików PDBx / mmCIF
Pliki PDB dystrybuowane przez Protein Data Bank mogą zawierać błędy formatowania, które powodują, że są niejednoznaczne lub trudne do przeanalizowania. Moduł Bio.PDB próbuje automatycznie radzić sobie z tymi błędami.
Moduł Bio.PDB implementuje dwa różne parsery, jeden w formacie mmCIF, a drugi w formacie pdb.
Nauczmy się szczegółowo analizować każdy z formatów -
Parser mmCIF
Pobierzmy przykładową bazę danych w formacie mmCIF z serwera pdb za pomocą poniższego polecenia -
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'mmCif')Spowoduje to pobranie określonego pliku (2fat.cif) z serwera i zapisanie go w bieżącym katalogu roboczym.
Tutaj PDBList zapewnia opcje wyświetlania i pobierania plików z internetowego serwera FTP PDB. metoda retrieve_pdb_file wymaga nazwy pliku do pobrania bez rozszerzenia. retrieve_pdb_file ma również opcję określenia katalogu pobierania, katalogu pdir i formatu pliku, format_pliku. Możliwe wartości formatu pliku są następujące -
- „MmCif” (domyślnie, plik PDBx / mmCif)
- „Pdb” (format PDB)
- „Xml” (format PMDML / XML)
- „Mmtf” (mocno skompresowany)
- „Pakiet” (archiwum w formacie PDB dla dużych struktur)
Aby załadować plik cif, użyj Bio.MMCIF.MMCIFParser, jak określono poniżej -
>>> parser = MMCIFParser(QUIET = True)
>>> data = parser.get_structure("2FAT", "2FAT.cif")Tutaj QUIET pomija ostrzeżenie podczas analizowania pliku. get_structure will parse the file and return the structure with id as 2FAT (pierwszy argument).
Po uruchomieniu powyższego polecenia analizuje plik i wyświetla ewentualne ostrzeżenie, jeśli jest dostępne.
Teraz sprawdź strukturę za pomocą poniższego polecenia -
>>> data
<Structure id = 2FAT>
To get the type, use type method as specified below,
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Pomyślnie przeanalizowaliśmy plik i uzyskaliśmy strukturę białka. Dowiemy się szczegółów struktury białka i jak ją uzyskać w następnym rozdziale.
Parser PDB
Pobierzmy przykładową bazę danych w formacie PDB z serwera pdb za pomocą poniższego polecenia -
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'pdb')Spowoduje to pobranie określonego pliku (pdb2fat.ent) z serwera i zapisanie go w bieżącym katalogu roboczym.
Aby załadować plik pdb, użyj Bio.PDB.PDBParser, jak określono poniżej -
>>> parser = PDBParser(PERMISSIVE = True, QUIET = True)
>>> data = parser.get_structure("2fat","pdb2fat.ent")Tutaj get_structure jest podobne do MMCIFParser. PERMISSIVE spróbuj przeanalizować dane białka tak elastycznie, jak to możliwe.
Teraz sprawdź strukturę i jej typ za pomocą fragmentu kodu podanego poniżej -
>>> data
<Structure id = 2fat>
>>> print(type(data))
<class 'Bio.PDB.Structure.Structure'>Cóż, struktura nagłówka przechowuje informacje ze słownika. Aby to zrobić, wpisz poniższe polecenie -
>>> print(data.header.keys()) dict_keys([
'name', 'head', 'deposition_date', 'release_date', 'structure_method', 'resolution',
'structure_reference', 'journal_reference', 'author', 'compound', 'source',
'keywords', 'journal'])
>>>Aby uzyskać nazwę, użyj następującego kodu -
>>> print(data.header["name"])
an anti-urokinase plasminogen activator receptor (upar) antibody: crystal
structure and binding epitope
>>>Możesz również sprawdzić datę i rozdzielczość za pomocą poniższego kodu -
>>> print(data.header["release_date"]) 2006-11-14
>>> print(data.header["resolution"]) 1.77Struktura PDB
Struktura PDB składa się z jednego modelu zawierającego dwa łańcuchy.
- łańcuch L, zawierający szereg reszt
- łańcuch H, zawierający szereg reszt
Każda reszta składa się z wielu atomów, z których każdy ma pozycję 3D reprezentowaną przez współrzędne (x, y, z).
Nauczmy się szczegółowo, jak uzyskać strukturę atomu w poniższej sekcji -
Model
Metoda Structure.get_models () zwraca iterator po modelach. Jest zdefiniowany poniżej -
>>> model = data.get_models()
>>> model
<generator object get_models at 0x103fa1c80>
>>> models = list(model)
>>> models [<Model id = 0>]
>>> type(models[0])
<class 'Bio.PDB.Model.Model'>Tutaj Model opisuje dokładnie jedną konformację 3D. Zawiera jeden lub więcej łańcuchów.
Łańcuch
Metoda Model.get_chain () zwraca iterator po łańcuchach. Jest zdefiniowany poniżej -
>>> chains = list(models[0].get_chains())
>>> chains
[<Chain id = L>, <Chain id = H>]
>>> type(chains[0])
<class 'Bio.PDB.Chain.Chain'>Tutaj Chain opisuje właściwą strukturę polipeptydu, tj. Kolejną sekwencję związanych reszt.
Pozostałość
Metoda Chain.get_residues () zwraca iterator po resztach. Jest zdefiniowany poniżej -
>>> residue = list(chains[0].get_residues())
>>> len(residue)
293
>>> residue1 = list(chains[1].get_residues())
>>> len(residue1)
311Cóż, Residue zawiera atomy należące do aminokwasu.
Atomy
Residue.get_atom () zwraca iterator po atomach, jak zdefiniowano poniżej -
>>> atoms = list(residue[0].get_atoms())
>>> atoms
[<Atom N>, <Atom CA>, <Atom C>, <Atom Ov, <Atom CB>, <Atom CG>, <Atom OD1>, <Atom OD2>]Atom zawiera trójwymiarową współrzędną atomu i nazywa się go wektorem. Jest to zdefiniowane poniżej
>>> atoms[0].get_vector()
<Vector 18.49, 73.26, 44.16>Reprezentuje wartości współrzędnych x, y i z.
Motyw sekwencji to wzór sekwencji nukleotydów lub aminokwasów. Motywy sekwencji są tworzone przez trójwymiarowe rozmieszczenie aminokwasów, które mogą nie sąsiadować ze sobą. Biopython zapewnia oddzielny moduł, Bio.motifs, aby uzyskać dostęp do funkcjonalności motywu sekwencji, jak określono poniżej -
from Bio import motifsTworzenie prostego motywu DNA
Stwórzmy prostą sekwencję motywów DNA za pomocą poniższego polecenia -
>>> from Bio import motifs
>>> from Bio.Seq import Seq
>>> DNA_motif = [ Seq("AGCT"),
... Seq("TCGA"),
... Seq("AACT"),
... ]
>>> seq = motifs.create(DNA_motif)
>>> print(seq) AGCT TCGA AACTAby policzyć wartości sekwencji, użyj poniższego polecenia -
>>> print(seq.counts)
0 1 2 3
A: 2.00 1.00 0.00 1.00
C: 0.00 1.00 2.00 0.00
G: 0.00 1.00 1.00 0.00
T: 1.00 0.00 0.00 2.00Użyj poniższego kodu, aby policzyć „A” w sekwencji -
>>> seq.counts["A", :]
(2, 1, 0, 1)Jeśli chcesz uzyskać dostęp do kolumn liczebności, użyj poniższego polecenia -
>>> seq.counts[:, 3]
{'A': 1, 'C': 0, 'T': 2, 'G': 0}Tworzenie logo sekwencji
Omówimy teraz, jak stworzyć logo sekwencji.
Rozważ poniższą sekwencję -
AGCTTACG
ATCGTACC
TTCCGAAT
GGTACGTA
AAGCTTGGMożesz stworzyć własne logo, korzystając z poniższego linku - http://weblogo.berkeley.edu/
Dodaj powyższą sekwencję i utwórz nowe logo i zapisz obraz o nazwie seq.png w swoim folderze biopython.
seq.pngPo utworzeniu obrazu uruchom teraz następujące polecenie -
>>> seq.weblogo("seq.png")Ten motyw sekwencji DNA jest reprezentowany jako logo sekwencji dla motywu wiążącego LexA.
Baza danych JASPAR
JASPAR to jedna z najpopularniejszych baz danych. Zapewnia funkcje dowolnego z formatów motywów do czytania, pisania i skanowania sekwencji. Przechowuje metainformacje dla każdego motywu.The module Bio.motifs contains a specialized class jaspar.Motif to represent meta-information attributes.
Ma następujące godne uwagi typy atrybutów -
- matrix_id - Unikalny identyfikator motywu JASPAR
- name - nazwa motywu
- tf_family - rodzina motywów, np. „Helix-Loop-Helix”
- data_type - typ danych używanych w motywie.
Utwórzmy format serwisów JASPAR o nazwie w sample.sites w folderze biopython. Jest zdefiniowany poniżej -
sample.sites
>MA0001 ARNT 1
AACGTGatgtccta
>MA0001 ARNT 2
CAGGTGggatgtac
>MA0001 ARNT 3
TACGTAgctcatgc
>MA0001 ARNT 4
AACGTGacagcgct
>MA0001 ARNT 5
CACGTGcacgtcgt
>MA0001 ARNT 6
cggcctCGCGTGcW powyższym pliku stworzyliśmy instancje motywów. Teraz stwórzmy obiekt motywu z powyższych instancji -
>>> from Bio import motifs
>>> with open("sample.sites") as handle:
... data = motifs.read(handle,"sites")
...
>>> print(data)
TF name None
Matrix ID None
Matrix:
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00Tutaj dane odczytują wszystkie instancje motywów z pliku sample.sites.
Aby wydrukować wszystkie instancje z danych, użyj poniższego polecenia -
>>> for instance in data.instances:
... print(instance)
...
AACGTG
CAGGTG
TACGTA
AACGTG
CACGTG
CGCGTGUżyj poniższego polecenia, aby policzyć wszystkie wartości -
>>> print(data.counts)
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00
>>>BioSQLto ogólny schemat bazy danych przeznaczony głównie do przechowywania sekwencji i powiązanych z nimi danych dla wszystkich silników RDBMS. Został zaprojektowany w taki sposób, że przechowuje dane ze wszystkich popularnych baz bioinformatycznych, takich jak GenBank, Swissport itp. Może być również używany do przechowywania danych wewnętrznych.
BioSQL obecnie zapewnia określony schemat dla poniższych baz danych -
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Zapewnia również minimalne wsparcie dla baz danych HSQLDB i Derby opartych na języku Java.
BioPython zapewnia bardzo proste, łatwe i zaawansowane możliwości ORM do pracy z bazą danych opartą na BioSQL. BioPython provides a module, BioSQL wykonać następującą funkcjonalność -
- Utwórz / usuń bazę danych BioSQL
- Połącz się z bazą danych BioSQL
- Przeanalizuj bazę danych sekwencji, taką jak GenBank, Swisport, wynik BLAST, wynik Entrez itp., I załaduj ją bezpośrednio do bazy danych BioSQL
- Pobierz dane sekwencji z bazy danych BioSQL
- Pobierz dane taksonomii z NCBI BLAST i przechowuj je w bazie danych BioSQL
- Uruchom dowolne zapytanie SQL w bazie danych BioSQL
Omówienie schematu bazy danych BioSQL
Zanim zagłębimy się w BioSQL, pozwól nam zrozumieć podstawy schematu BioSQL. Schemat BioSQL zapewnia ponad 25 tabel do przechowywania danych sekwencji, funkcji sekwencji, kategorii / ontologii sekwencji i informacji taksonomicznych. Oto niektóre z ważnych tabel -
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
Tworzenie bazy danych BioSQL
W tej sekcji stwórzmy przykładową bazę danych BioSQL, biosql przy użyciu schematu dostarczonego przez zespół BioSQL. Będziemy pracować z bazą danych SQLite, ponieważ jest naprawdę łatwa do rozpoczęcia i nie ma skomplikowanej konfiguracji.
W tym miejscu utworzymy bazę danych BioSQL opartą na SQLite, wykonując poniższe kroki.
Step 1 - Pobierz silnik bazy danych SQLite i zainstaluj go.
Step 2 - Pobierz projekt BioSQL z adresu URL GitHub. https://github.com/biosql/biosql
Step 3 - Otwórz konsolę i utwórz katalog za pomocą mkdir i wejdź do niego.
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosqlStep 4 - Uruchom poniższe polecenie, aby utworzyć nową bazę danych SQLite.
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>Step 5 - Skopiuj plik biosqldb-sqlite.sql z projektu BioSQL (/ sql / biosqldb-sqlite.sql`) i zapisz go w bieżącym katalogu.
Step 6 - Uruchom poniższe polecenie, aby utworzyć wszystkie tabele.
sqlite> .read biosqldb-sqlite.sqlTeraz wszystkie tabele są tworzone w naszej nowej bazie danych.
Step 7 - Uruchom poniższe polecenie, aby wyświetlić wszystkie nowe tabele w naszej bazie danych.
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>Pierwsze trzy polecenia to polecenia konfiguracyjne służące do konfigurowania programu SQLite w celu wyświetlenia wyniku w sformatowany sposób.
Step 8 - Skopiuj przykładowy plik GenBank, ls_orchid.gbk dostarczony przez zespół BioPython https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk do bieżącego katalogu i zapisz go jako orchid.gbk.
Step 9 - Utwórz skrypt w Pythonie, load_orchid.py, używając poniższego kodu i wykonaj go.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Powyższy kod analizuje rekord w pliku i konwertuje go na obiekty Pythona i wstawia do bazy danych BioSQL. Przeanalizujemy kod w dalszej części.
Na koniec stworzyliśmy nową bazę danych BioSQL i załadowaliśmy do niej kilka przykładowych danych. W następnym rozdziale omówimy ważne tabele.
Prosty schemat ER
biodatabase tabela znajduje się na szczycie hierarchii, a jej głównym celem jest zorganizowanie zestawu danych sekwencyjnych w pojedynczą grupę / wirtualną bazę danych. Every entry in the biodatabase refers to a separate database and it does not mingle with another database. Wszystkie powiązane tabele w bazie danych BioSQL zawierają odniesienia do wpisu bazy danych biologicznych.
bioentrytabela zawiera wszystkie szczegóły dotyczące sekwencji z wyjątkiem danych sekwencji. dane sekwencyjne konkretnegobioentry będą przechowywane w biosequence stół.
Takson i nazwa_takonu są szczegółami taksonomii i każda pozycja odsyła do tej tabeli w celu określenia informacji o taksonu.
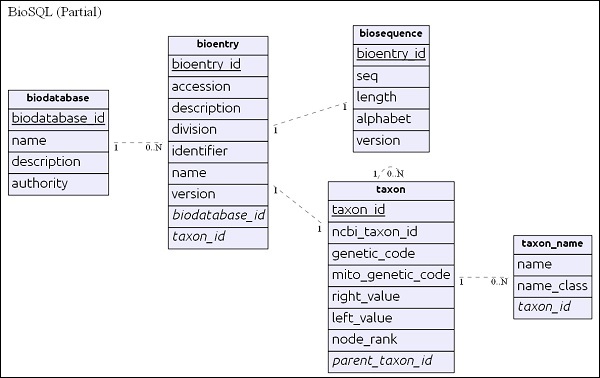
Po zrozumieniu schematu przyjrzyjmy się niektórym zapytaniom w następnej sekcji.
Zapytania BioSQL
Zagłębmy się w niektóre zapytania SQL, aby lepiej zrozumieć, w jaki sposób dane są zorganizowane i tabele są ze sobą powiązane. Zanim przejdziesz dalej, otwórzmy bazę danych za pomocą poniższego polecenia i ustaw kilka poleceń formatujących -
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns.header and .mode are formatting options to better visualize the data. Możesz również użyć dowolnego edytora SQLite do uruchomienia zapytania.
Wymień wirtualną bazę danych sekwencji dostępną w systemie, jak podano poniżej -
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>Tutaj mamy tylko jedną bazę danych, orchid.
Wypisz wpisy (pierwsze 3) dostępne w bazie danych orchid z poniższym kodem
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>Wymień szczegóły sekwencji związane z wpisem (przystąpienie - Z78530, nazwa - gen C. fasciculatum 5.8S rRNA oraz DNA ITS1 i ITS2) z podanym kodem -
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>Uzyskaj pełną sekwencję związaną z wpisem (dostęp - Z78530, nazwa - gen C. fasciculatum 5.8S rRNA oraz DNA ITS1 i ITS2), używając poniższego kodu -
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>Lista taksonów powiązanych z bio bazą danych, orchidea
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>Załaduj dane do bazy danych BioSQL
W tym rozdziale nauczmy się, jak ładować dane sekwencji do bazy danych BioSQL. Mamy już kod do ładowania danych do bazy danych w poprzedniej sekcji, a kod wygląda następująco -
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()Przyjrzymy się dokładniej każdej linii kodu i jego przeznaczeniu -
Line 1 - Ładuje moduł SeqIO.
Line 2- Ładuje moduł BioSeqDatabase. Ten moduł zapewnia wszystkie funkcje do interakcji z bazą danych BioSQL.
Line 3 - Ładuje moduł systemu operacyjnego.
Line 5- open_database otwiera określoną bazę danych (db) ze skonfigurowanym sterownikiem (sterownikiem) i zwraca uchwyt do bazy danych BioSQL (serwera). Biopython obsługuje bazy danych sqlite, mysql, postgresql i oracle.
Line 6-10- metoda load_database_sql ładuje sql z zewnętrznego pliku i wykonuje go. Metoda commit zatwierdza transakcję. Możemy pominąć ten krok, ponieważ utworzyliśmy już bazę danych ze schematem.
Line 12 - metody new_database tworzy nową wirtualną bazę danych, orchid i zwraca db uchwyt do wykonania polecenia w bazie danych orchidei.
Line 13- metoda ładowania ładuje wpisy sekwencji (iterowalny SeqRecord) do bazy danych orchidei. SqlIO.parse analizuje bazę danych GenBank i zwraca wszystkie zawarte w niej sekwencje jako iterowalne SeqRecord. Drugi parametr (True) metody load nakazuje jej pobranie szczegółów taksonomii danych sekwencji ze strony internetowej NCBI, jeśli nie są one jeszcze dostępne w systemie.
Line 14 - commit zatwierdza transakcję.
Line 15 - close zamyka połączenie z bazą danych i niszczy uchwyt serwera.
Pobierz dane sekwencji
Pobierzmy sekwencję z identyfikatorem 2765658 z bazy danych orchidei, jak poniżej -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))Tutaj serwer ["orchid"] zwraca uchwyt do pobierania danych z wirtualnej bazy danychorchid. lookup metoda zapewnia opcję wyboru sekwencji na podstawie kryteriów i wybraliśmy sekwencję o identyfikatorze 2765658. lookupzwraca informacje o sekwencji jako SeqRecordobject. Ponieważ wiemy już, jak pracować z SeqRecord`, łatwo jest uzyskać z niego dane.
Usuń bazę danych
Usunięcie bazy danych jest tak proste, jak wywołanie metody remove_database z odpowiednią nazwą bazy danych, a następnie zatwierdzenie jej zgodnie z poniższym opisem -
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()Genetyka populacji odgrywa ważną rolę w teorii ewolucji. Analizuje różnice genetyczne między gatunkami, a także dwoma lub więcej osobnikami w ramach tego samego gatunku.
Biopython dostarcza moduł Bio.PopGen do genetyki populacyjnej i obsługuje głównie `GenePop, popularny pakiet genetyki opracowany przez Michela Raymonda i Francois Rousset.
Prosty parser
Napiszmy prostą aplikację do analizowania formatu GenePop i zrozumienia koncepcji.
Pobierz plik genePop dostarczony przez zespół Biopython z linku podanego poniżej -https://raw.githubusercontent.com/biopython/biopython/master/Tests/PopGen/c3line.gen
Załaduj moduł GenePop za pomocą poniższego fragmentu kodu -
from Bio.PopGen import GenePopPrzeanalizuj plik za pomocą metody GenePop.read, jak poniżej -
record = GenePop.read(open("c3line.gen"))Pokaż loci i informacje o populacji, jak podano poniżej -
>>> record.loci_list
['136255903', '136257048', '136257636']
>>> record.pop_list
['4', 'b3', '5']
>>> record.populations
[[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (3, 4), (2, 2)]),
('3', [(3, 3), (4, 4), (2, 2)]), ('4', [(3, 3), (4, 3), (None, None)])],
[('b1', [(None, None), (4, 4), (2, 2)]), ('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]), ('4',
[(None, None), (4, 4), (2, 2)]), ('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Tutaj w pliku dostępne są trzy loci i trzy zestawy populacji: populacja pierwsza ma 4 rekordy, populacja druga ma 3 rekordy, a populacja trzecia 5 rekordów. record.populations pokazuje wszystkie zestawy populacji z danymi alleli dla każdego locus.
Manipuluj plikiem GenePop
Biopython zapewnia opcje usuwania danych dotyczących lokalizacji i populacji.
Remove a population set by position,
>>> record.remove_population(0)
>>> record.populations
[[('b1', [(None, None), (4, 4), (2, 2)]),
('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]),
('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]),
('4', [(None, None), (4, 4), (2, 2)]),
('5', [(3, 3), (4, 4), (2, 2)])]]
>>>Remove a locus by position,
>>> record.remove_locus_by_position(0)
>>> record.loci_list
['136257048', '136257636']
>>> record.populations
[[('b1', [(4, 4), (2, 2)]), ('b2', [(4, 4), (2, 2)]), ('b3', [(4, 4), (2, 2)])],
[('1', [(4, 4), (2, 2)]), ('2', [(1, 4), (2, 2)]),
('3', [(1, 1), (2, 2)]), ('4', [(4, 4), (2, 2)]), ('5', [(4, 4), (2, 2)])]]
>>>Remove a locus by name,
>>> record.remove_locus_by_name('136257636') >>> record.loci_list
['136257048']
>>> record.populations
[[('b1', [(4, 4)]), ('b2', [(4, 4)]), ('b3', [(4, 4)])],
[('1', [(4, 4)]), ('2', [(1, 4)]),
('3', [(1, 1)]), ('4', [(4, 4)]), ('5', [(4, 4)])]]
>>>Interfejs z oprogramowaniem GenePop
Biopython zapewnia interfejsy do interakcji z oprogramowaniem GenePop, a tym samym udostępnia z niego wiele funkcji. Służy do tego moduł Bio.PopGen.GenePop. Jednym z takich łatwych w użyciu interfejsów jest EasyController. Sprawdźmy, jak przeanalizować plik GenePop i przeprowadzić analizę za pomocą EasyController.
Najpierw zainstaluj oprogramowanie GenePop i umieść folder instalacyjny w ścieżce systemowej. Aby uzyskać podstawowe informacje o pliku GenePop, utwórz obiekt EasyController, a następnie wywołaj metodę get_basic_info, jak określono poniżej -
>>> from Bio.PopGen.GenePop.EasyController import EasyController
>>> ec = EasyController('c3line.gen')
>>> print(ec.get_basic_info())
(['4', 'b3', '5'], ['136255903', '136257048', '136257636'])
>>>Tutaj pierwsza pozycja to lista populacji, a druga to lista miejsc.
Aby uzyskać listę wszystkich alleli określonego locus, wywołaj metodę get_alleles_all_pops, przekazując nazwę miejsca, jak określono poniżej -
>>> allele_list = ec.get_alleles_all_pops("136255903")
>>> print(allele_list)
[2, 3]Aby uzyskać listę alleli według określonej populacji i locus, wywołaj get_alleles, przekazując nazwę miejsca i pozycję populacji, jak podano poniżej -
>>> allele_list = ec.get_alleles(0, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(1, "136255903")
>>> print(allele_list)
[]
>>> allele_list = ec.get_alleles(2, "136255903")
>>> print(allele_list)
[2, 3]
>>>Podobnie EasyController udostępnia wiele funkcji: częstotliwość alleli, częstotliwość genotypów, statystyki multilocus F, równowaga Hardy'ego-Weinberga, równowaga sprzężeń itp.
Genom to kompletny zestaw DNA, w tym wszystkie jego geny. Analiza genomu odnosi się do badania poszczególnych genów i ich roli w dziedziczeniu.
Diagram genomu
Diagram genomu przedstawia informacje genetyczne w postaci wykresów. Biopython używa modułu Bio.Graphics.GenomeDiagram do reprezentowania GenomeDiagram. Moduł GenomeDiagram wymaga zainstalowania ReportLab.
Kroki tworzenia diagramu
Proces tworzenia diagramu zasadniczo przebiega według poniższego prostego wzoru -
Utwórz FeatureSet dla każdego oddzielnego zestawu funkcji, które chcesz wyświetlić, i dodaj do nich obiekty Bio.SeqFeature.
Utwórz zestaw wykresów dla każdego wykresu, który chcesz wyświetlić, i dodaj do nich dane wykresu.
Utwórz ścieżkę dla każdej ścieżki, którą chcesz na diagramie i dodaj GraphSets i FeatureSets do żądanych ścieżek.
Utwórz diagram i dodaj do niego ścieżki.
Powiedz Diagramowi, aby narysował obraz.
Zapisz obraz do pliku.
Weźmy przykład wejściowego pliku GenBank -
https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbki odczytaj rekordy z obiektu SeqRecord, a następnie narysuj diagram genomu. Jest to wyjaśnione poniżej,
Najpierw zaimportujemy wszystkie moduły, jak pokazano poniżej -
>>> from reportlab.lib import colors
>>> from reportlab.lib.units import cm
>>> from Bio.Graphics import GenomeDiagramTeraz zaimportuj moduł SeqIO, aby odczytać dane -
>>> from Bio import SeqIO
record = SeqIO.read("example.gb", "genbank")Tutaj rekord odczytuje sekwencję z pliku genbank.
Teraz utwórz pusty diagram, aby dodać ścieżkę i zestaw funkcji -
>>> diagram = GenomeDiagram.Diagram(
"Yersinia pestis biovar Microtus plasmid pPCP1")
>>> track = diagram.new_track(1, name="Annotated Features")
>>> feature = track.new_set()Teraz możemy zastosować zmiany motywu kolorystycznego, używając alternatywnych kolorów od zielonego do szarego, jak zdefiniowano poniżej -
>>> for feature in record.features:
>>> if feature.type != "gene":
>>> continue
>>> if len(feature) % 2 == 0:
>>> color = colors.blue
>>> else:
>>> color = colors.red
>>>
>>> feature.add_feature(feature, color=color, label=True)Teraz możesz zobaczyć poniższą odpowiedź na ekranie -
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dc90>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dfd0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x1007627d0>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57290>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57050>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57390>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57590>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57410>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57490>
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d574d0>Narysujmy diagram dla powyższych rekordów wejściowych -
>>> diagram.draw(
format = "linear", orientation = "landscape", pagesize = 'A4',
... fragments = 4, start = 0, end = len(record))
>>> diagram.write("orchid.pdf", "PDF")
>>> diagram.write("orchid.eps", "EPS")
>>> diagram.write("orchid.svg", "SVG")
>>> diagram.write("orchid.png", "PNG")Po wykonaniu powyższego polecenia możesz zobaczyć następujący obraz zapisany w twoim katalogu Biopython.
** Result **
genome.png
Możesz także narysować obraz w formacie okrągłym, wprowadzając poniższe zmiany -
>>> diagram.draw(
format = "circular", circular = True, pagesize = (20*cm,20*cm),
... start = 0, end = len(record), circle_core = 0.7)
>>> diagram.write("circular.pdf", "PDF")Przegląd chromosomów
Cząsteczka DNA jest upakowana w nitkowate struktury zwane chromosomami. Każdy chromosom składa się z DNA, wielokrotnie zwiniętego ciasno wokół białek zwanych histonami, które wspierają jego strukturę.
Chromosomy nie są widoczne w jądrze komórki - nawet pod mikroskopem - gdy komórka się nie dzieli. Jednak DNA, z którego składają się chromosomy, podczas podziału komórki staje się ściślej upakowane i jest następnie widoczne pod mikroskopem.
U ludzi każda komórka zwykle zawiera 23 pary chromosomów, w sumie 46. Dwadzieścia dwie z tych par, zwanych autosomami, wyglądają tak samo zarówno u mężczyzn, jak iu kobiet. Dwudziesta trzecia para, chromosomy płci, różni się między mężczyznami i kobietami. Kobiety mają dwie kopie chromosomu X, podczas gdy mężczyźni mają jeden chromosom X i jeden Y.
Fenotyp jest definiowany jako obserwowalny charakter lub cecha wykazywana przez organizm na tle określonej substancji chemicznej lub środowiska. Mikromacierz fenotypowa jednocześnie mierzy reakcję organizmu na większą liczbę chemikaliów i środowiska oraz analizuje dane w celu zrozumienia mutacji genów, cech genów itp.
Biopython zapewnia doskonały moduł, Bio.Phenotype, do analizy danych fenotypowych. Nauczmy się analizować, interpolować, wyodrębniać i analizować dane z mikromacierzy fenotypowej w tym rozdziale.
Rozbiór gramatyczny zdania
Dane z mikromacierzy fenotypowej mogą mieć dwa formaty: CSV i JSON. Biopython obsługuje oba formaty. Parser Biopython analizuje dane z mikromacierzy fenotypu i zwraca je jako zbiór obiektów PlateRecord. Każdy obiekt PlateRecord zawiera kolekcję obiektów WellRecord. Każdy obiekt WellRecord przechowuje dane w 8 wierszach i 12 kolumnach. Osiem rzędów są reprezentowane przez A do H i 12 kolumny są reprezentowane przez 01 do 12. Na przykład, 4 p rzędów i 6 p kolumny są reprezentowane przez D06.
Rozumiemy format i koncepcję analizowania na następującym przykładzie -
Step 1 - Pobierz plik Plates.csv dostarczony przez zespół Biopython - https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/Plates.csv
Step 2 - Załaduj moduł Phenotpe jak poniżej -
>>> from Bio import phenotypeStep 3- Wywołaj metodę phenotype.parse przekazując plik danych i opcję formatu („pm-csv”). Zwraca iterowalny PlateRecord, jak poniżej,
>>> plates = list(phenotype.parse('Plates.csv', "pm-csv"))
>>> plates
[PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'],WellRecord['A03'], ..., WellRecord['H12']')]
>>>Step 4 - Uzyskaj dostęp do pierwszej płyty z listy, jak poniżej -
>>> plate = plates[0]
>>> plate
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ...,
WellRecord['H12']')
>>>Step 5- Jak wspomniano wcześniej, płyta zawiera 8 rzędów, z których każdy ma 12 elementów. Dostęp do WellRecord można uzyskać na dwa sposoby, jak określono poniżej -
>>> well = plate["A04"]
>>> well = plate[0, 4]
>>> well WellRecord('(0.0, 0.0), (0.25, 0.0), (0.5, 0.0), (0.75, 0.0),
(1.0, 0.0), ..., (71.75, 388.0)')
>>>Step 6 - Każdy dołek będzie miał serię pomiarów w różnych punktach czasowych i można uzyskać do niego dostęp za pomocą pętli for, jak określono poniżej -
>>> for v1, v2 in well:
... print(v1, v2)
...
0.0 0.0
0.25 0.0
0.5 0.0
0.75 0.0
1.0 0.0
...
71.25 388.0
71.5 388.0
71.75 388.0
>>>Interpolacja
Interpolacja zapewnia lepszy wgląd w dane. Biopython zapewnia metody interpolacji danych WellRecord w celu uzyskania informacji o pośrednich punktach czasowych. Składnia jest podobna do indeksowania list i dlatego jest łatwa do nauczenia.
Aby uzyskać dane po 20,1 godzinie, po prostu przekaż jako wartości indeksu, jak określono poniżej -
>>> well[20.10]
69.40000000000003
>>>Możemy przekroczyć punkt początkowy i końcowy, jak również określono poniżej -
>>> well[20:30]
[67.0, 84.0, 102.0, 119.0, 135.0, 147.0, 158.0, 168.0, 179.0, 186.0]
>>>Powyższe polecenie interpoluje dane od 20 do 30 godzin z 1-godzinnym odstępem. Domyślnie interwał to 1 godzina i możemy go zmienić na dowolną wartość. Na przykład dajmy 15 minut (0,25 godziny) interwał, jak określono poniżej -
>>> well[20:21:0.25]
[67.0, 73.0, 75.0, 81.0]
>>>Przeanalizuj i wyodrębnij
Biopython zapewnia metodę odpowiednią do analizy danych WellRecord przy użyciu funkcji sigmoidalnych Gompertza, Logistic i Richardsa. Domyślnie metoda fit wykorzystuje funkcję Gompertz. Aby wykonać zadanie, musimy wywołać metodę dopasowania obiektu WellRecord. Kodowanie jest następujące -
>>> well.fit()
Traceback (most recent call last):
...
Bio.MissingPythonDependencyError: Install scipy to extract curve parameters.
>>> well.model
>>> getattr(well, 'min') 0.0
>>> getattr(well, 'max') 388.0
>>> getattr(well, 'average_height')
205.42708333333334
>>>Biopython jest zależny od modułu Scipy do wykonywania zaawansowanych analiz. Oblicza szczegóły min, max i Average_height bez użycia modułu scipy.
W tym rozdziale wyjaśniono, jak wykreślić sekwencje. Zanim przejdziemy do tego tematu, zrozumiemy podstawy kreślenia.
Konspiratorstwo
Matplotlib to biblioteka kreśląca w języku Python, która tworzy wartości liczbowe w różnych formatach. Możemy tworzyć różne typy wykresów, takie jak wykres liniowy, histogramy, wykres słupkowy, wykres kołowy, wykres punktowy itp.
pyLab is a module that belongs to the matplotlib which combines the numerical module numpy with the graphical plotting module pyplot.Biopython używa modułu pylab do kreślenia sekwencji. Aby to zrobić, musimy zaimportować poniższy kod -
import pylabPrzed importem musimy zainstalować pakiet matplotlib za pomocą polecenia pip z poleceniem podanym poniżej -
pip install matplotlibPrzykładowy plik wejściowy
Utwórz przykładowy plik o nazwie plot.fasta w katalogu Biopython i dodaj następujące zmiany -
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF
>seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME
>seq2 EEYQTWEEFARAAEKLYLTDPMKVRVVLKYRHCDGNLCMKVTDDAVCLQYKTDQAQDVKKVEKLHGK
>seq3 MYQVWEEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVCLQYKTDQAQDV
>seq4 EEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVVSYEMRLFGVQKDNFALEHSLL
>seq5 SWEEFAKAAEVLYLEDPMKCRMCTKYRHVDHKLVVKLTDNHTVLKYVTDMAQDVKKIEKLTTLLMR
>seq6 FTNWEEFAKAAERLHSANPEKCRFVTKYNHTKGELVLKLTDDVVCLQYSTNQLQDVKKLEKLSSTLLRSI
>seq7 SWEEFVERSVQLFRGDPNATRYVMKYRHCEGKLVLKVTDDRECLKFKTDQAQDAKKMEKLNNIFF
>seq8 SWDEFVDRSVQLFRADPESTRYVMKYRHCDGKLVLKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM
>seq9 KNWEDFEIAAENMYMANPQNCRYTMKYVHSKGHILLKMSDNVKCVQYRAENMPDLKK
>seq10 FDSWDEFVSKSVELFRNHPDTTRYVVKYRHCEGKLVLKVTDNHECLKFKTDQAQDAKKMEKDziałka liniowa
Teraz stwórzmy prosty wykres liniowy dla powyższego pliku fasta.
Step 1 - Importuj moduł SeqIO do odczytu pliku fasta.
>>> from Bio import SeqIOStep 2 - Przeanalizuj plik wejściowy.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Zaimportujmy moduł pylab.
>>> import pylabStep 4 - Skonfiguruj wykres liniowy, przypisując etykiety osi X i Y.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Skonfiguruj wykres liniowy, ustawiając wyświetlanie siatki.
>>> pylab.grid()Step 6 - Narysuj prosty wykres liniowy, wywołując metodę plot i podając rekordy jako dane wejściowe.
>>> pylab.plot(records)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Na koniec zapisz wykres za pomocą poniższego polecenia.
>>> pylab.savefig("lines.png")Wynik
Po wykonaniu powyższego polecenia możesz zobaczyć następujący obraz zapisany w twoim katalogu Biopython.
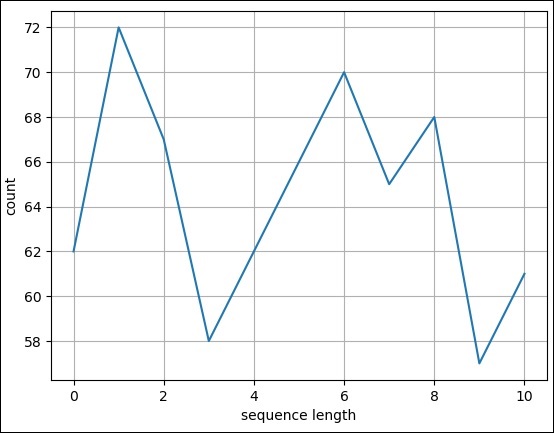
Wykres histogramu
Histogram jest używany w przypadku danych ciągłych, gdzie przedziały reprezentują zakresy danych. Rysowanie histogramu jest takie samo jak wykresu liniowego, z wyjątkiem pylab.plot. Zamiast tego wywołaj metodę hist modułu pylab z rekordami i pewną wartością niestandardową dla pojemników (5). Pełne kodowanie jest następujące -
Step 1 - Importuj moduł SeqIO do odczytu pliku fasta.
>>> from Bio import SeqIOStep 2 - Przeanalizuj plik wejściowy.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57Step 3 - Zaimportujmy moduł pylab.
>>> import pylabStep 4 - Skonfiguruj wykres liniowy, przypisując etykiety osi X i Y.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>Step 5 - Skonfiguruj wykres liniowy, ustawiając wyświetlanie siatki.
>>> pylab.grid()Step 6 - Narysuj prosty wykres liniowy, wywołując metodę plot i podając rekordy jako dane wejściowe.
>>> pylab.hist(records,bins=5)
(array([2., 3., 1., 3., 2.]), array([57., 60., 63., 66., 69., 72.]), <a list
of 5 Patch objects>)
>>>Step 7 - Na koniec zapisz wykres za pomocą poniższego polecenia.
>>> pylab.savefig("hist.png")Wynik
Po wykonaniu powyższego polecenia możesz zobaczyć następujący obraz zapisany w twoim katalogu Biopython.
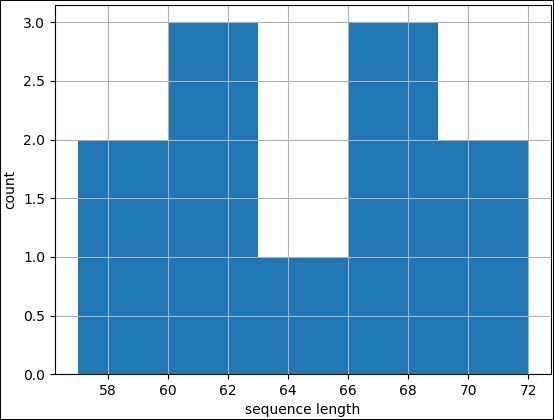
Procent GC w sekwencji
Procent GC jest jednym z powszechnie stosowanych danych analitycznych do porównywania różnych sekwencji. Możemy zrobić prosty wykres liniowy za pomocą GC Percentage zbioru sekwencji i od razu go porównać. Tutaj możemy po prostu zmienić dane z długości sekwencji na procent GC. Pełne kodowanie podano poniżej -
Step 1 - Importuj moduł SeqIO do odczytu pliku fasta.
>>> from Bio import SeqIOStep 2 - Przeanalizuj plik wejściowy.
>>> from Bio.SeqUtils import GC
>>> gc = sorted(GC(rec.seq) for rec in SeqIO.parse("plot.fasta", "fasta"))Step 3 - Zaimportujmy moduł pylab.
>>> import pylabStep 4 - Skonfiguruj wykres liniowy, przypisując etykiety osi X i Y.
>>> pylab.xlabel("Genes")
Text(0.5, 0, 'Genes')
>>> pylab.ylabel("GC Percentage")
Text(0, 0.5, 'GC Percentage')
>>>Step 5 - Skonfiguruj wykres liniowy, ustawiając wyświetlanie siatki.
>>> pylab.grid()Step 6 - Narysuj prosty wykres liniowy, wywołując metodę plot i podając rekordy jako dane wejściowe.
>>> pylab.plot(gc)
[<matplotlib.lines.Line2D object at 0x10b6869d 0>]Step 7 - Na koniec zapisz wykres za pomocą poniższego polecenia.
>>> pylab.savefig("gc.png")Wynik
Po wykonaniu powyższego polecenia możesz zobaczyć następujący obraz zapisany w twoim katalogu Biopython.
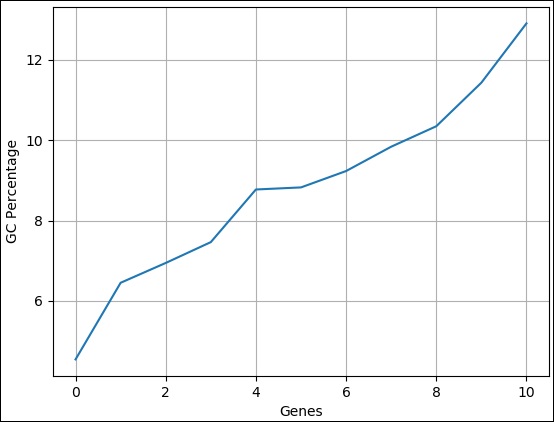
Ogólnie rzecz biorąc, Analiza skupień polega na grupowaniu zestawu obiektów w tej samej grupie. Pojęcie to jest wykorzystywane głównie w eksploracji danych, statystycznej analizie danych, uczeniu maszynowym, rozpoznawaniu wzorców, analizie obrazu, bioinformatyce itp. Można to osiągnąć za pomocą różnych algorytmów, aby zrozumieć, w jaki sposób klaster jest szeroko stosowany w różnych analizach.
Według Bioinformatics analiza skupień jest stosowana głównie w analizie danych dotyczących ekspresji genów w celu znalezienia grup genów o podobnej ekspresji genów.
W tym rozdziale sprawdzimy ważne algorytmy w Biopythonie, aby zrozumieć podstawy klastrowania na rzeczywistym zbiorze danych.
Biopython używa modułu Bio.Cluster do implementacji wszystkich algorytmów. Obsługuje następujące algorytmy -
- Klastrowanie hierarchiczne
- K - Klastrowanie
- Mapy samoorganizujące się
- Analiza głównych składowych
Przedstawmy krótkie wprowadzenie do powyższych algorytmów.
Klastrowanie hierarchiczne
Klastrowanie hierarchiczne jest używane do łączenia każdego węzła na podstawie miary odległości z najbliższym sąsiadem i tworzenia klastra. Węzeł Bio.Cluster ma trzy atrybuty: lewy, prawy i odległość. Utwórzmy prosty klaster, jak pokazano poniżej -
>>> from Bio.Cluster import Node
>>> n = Node(1,10)
>>> n.left = 11
>>> n.right = 0
>>> n.distance = 1
>>> print(n)
(11, 0): 1Jeśli chcesz zbudować klastrowanie oparte na drzewie, użyj poniższego polecenia -
>>> n1 = [Node(1, 2, 0.2), Node(0, -1, 0.5)] >>> n1_tree = Tree(n1)
>>> print(n1_tree)
(1, 2): 0.2
(0, -1): 0.5
>>> print(n1_tree[0])
(1, 2): 0.2Zróbmy hierarchiczne grupowanie za pomocą modułu Bio.Cluster.
Rozważmy, że odległość jest zdefiniowana w tablicy.
>>> import numpy as np
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Teraz dodaj tablicę odległości w grupie drzew.
>>> from Bio.Cluster import treecluster
>>> cluster = treecluster(distance)
>>> print(cluster)
(2, 1): 0.666667
(-1, 0): 9.66667Powyższa funkcja zwraca obiekt klastra Tree. Ten obiekt zawiera węzły, w których liczba elementów jest zgrupowana jako wiersze lub kolumny.
K - Klastrowanie
Jest to rodzaj algorytmu partycjonowania, który dzieli się na k - średnie, mediany i skupienia medoidów. Przyjrzyjmy się pokrótce każdemu z klastrów.
Grupowanie środków K
Takie podejście jest popularne w eksploracji danych. Celem tego algorytmu jest znalezienie grup w danych, przy czym liczbę grup reprezentuje zmienna K.
Algorytm działa iteracyjnie, aby przypisać każdy punkt danych do jednej z grup K na podstawie dostarczonych funkcji. Punkty danych są grupowane na podstawie podobieństwa cech.
>>> from Bio.Cluster import kcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid, error,found = kcluster(data)
>>> print(clusterid) [0 0 1]
>>> print(found)
1Klastry median K
Jest to inny typ algorytmu grupowania, który oblicza średnią dla każdego klastra w celu określenia jego środka ciężkości.
Grupowanie K-medoidów
Podejście to opiera się na zadanym zestawie elementów, wykorzystując macierz odległości i liczbę klastrów przekazanych przez użytkownika.
Rozważ macierz odległości, jak określono poniżej -
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])Możemy obliczyć grupowanie k-medoidów za pomocą poniższego polecenia -
>>> from Bio.Cluster import kmedoids
>>> clusterid, error, found = kmedoids(distance)Rozważmy przykład.
Funkcja kcluster przyjmuje macierz danych jako dane wejściowe, a nie instancje Seq. Musisz przekonwertować swoje sekwencje na macierz i dostarczyć to do funkcji kcluster.
Jednym ze sposobów konwersji danych do macierzy zawierającej tylko elementy numeryczne jest użycie rozszerzenia numpy.fromstringfunkcjonować. Zasadniczo tłumaczy każdą literę w sekwencji na jej odpowiednik w ASCII.
Tworzy to dwuwymiarową tablicę zakodowanych sekwencji, którą funkcja kcluster rozpoznała i wykorzystuje do grupowania sekwencji.
>>> from Bio.Cluster import kcluster
>>> import numpy as np
>>> sequence = [ 'AGCT','CGTA','AAGT','TCCG']
>>> matrix = np.asarray([np.fromstring(s, dtype=np.uint8) for s in sequence])
>>> clusterid,error,found = kcluster(matrix)
>>> print(clusterid) [1 0 0 1]Mapy samoorganizujące się
To podejście jest rodzajem sztucznej sieci neuronowej. Jest rozwijany przez Kohonena i często nazywany mapą Kohonena. Organizuje elementy w klastry w oparciu o topologię prostokątną.
Utwórzmy prosty klaster, używając tej samej odległości tablicy, jak pokazano poniżej -
>>> from Bio.Cluster import somcluster
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> clusterid,map = somcluster(data)
>>> print(map)
[[[-1.36032469 0.38667395]]
[[-0.41170578 1.35295911]]]
>>> print(clusterid)
[[1 0]
[1 0]
[1 0]]Tutaj, clusterid to tablica z dwiema kolumnami, w której liczba wierszy jest równa liczbie elementów, które zostały zgrupowane, a data jest tablicą zawierającą wymiary wierszy lub kolumn.
Analiza głównych składowych
Analiza głównych komponentów jest przydatna do wizualizacji danych wielowymiarowych. Jest to metoda, która wykorzystuje proste operacje na macierzach z algebry liniowej i statystyki do obliczenia rzutu oryginalnych danych na tę samą liczbę lub mniej wymiarów.
Principal Component Analysis zwraca kolumnę krotki, współrzędne, składniki i wartości własne. Przyjrzyjmy się podstawom tej koncepcji.
>>> from numpy import array
>>> from numpy import mean
>>> from numpy import cov
>>> from numpy.linalg import eig
# define a matrix
>>> A = array([[1, 2], [3, 4], [5, 6]])
>>> print(A)
[[1 2]
[3 4]
[5 6]]
# calculate the mean of each column
>>> M = mean(A.T, axis = 1)
>>> print(M)
[ 3. 4.]
# center columns by subtracting column means
>>> C = A - M
>>> print(C)
[[-2. -2.]
[ 0. 0.]
[ 2. 2.]]
# calculate covariance matrix of centered matrix
>>> V = cov(C.T)
>>> print(V)
[[ 4. 4.]
[ 4. 4.]]
# eigendecomposition of covariance matrix
>>> values, vectors = eig(V)
>>> print(vectors)
[[ 0.70710678 -0.70710678]
[ 0.70710678 0.70710678]]
>>> print(values)
[ 8. 0.]Zastosujmy te same prostokątne dane macierzy do modułu Bio.Cluster, jak zdefiniowano poniżej -
>>> from Bio.Cluster import pca
>>> from numpy import array
>>> data = array([[1, 2], [3, 4], [5, 6]])
>>> columnmean, coordinates, components, eigenvalues = pca(data)
>>> print(columnmean)
[ 3. 4.]
>>> print(coordinates)
[[-2.82842712 0. ]
[ 0. 0. ]
[ 2.82842712 0. ]]
>>> print(components)
[[ 0.70710678 0.70710678]
[ 0.70710678 -0.70710678]]
>>> print(eigenvalues)
[ 4. 0.]Bioinformatyka to doskonały obszar do zastosowania algorytmów uczenia maszynowego. Mamy tutaj informacje genetyczne dużej liczby organizmów i nie jest możliwe ręczne przeanalizowanie wszystkich tych informacji. Jeśli zastosujemy odpowiedni algorytm uczenia maszynowego, możemy z tych danych wydobyć wiele przydatnych informacji. Biopython zapewnia użyteczny zestaw algorytmów do nadzorowanego uczenia maszynowego.
Uczenie nadzorowane jest oparte na zmiennej wejściowej (X) i zmiennej wyjściowej (Y). Wykorzystuje algorytm, aby nauczyć się funkcji mapowania z wejścia do wyjścia. Jest zdefiniowany poniżej -
Y = f(X)Głównym celem tego podejścia jest przybliżenie funkcji mapowania, a gdy masz nowe dane wejściowe (x), możesz przewidzieć zmienne wyjściowe (Y) dla tych danych.
Model regresji logistycznej
Regresja logistyczna to nadzorowany algorytm uczenia maszynowego. Służy do znalezienia różnicy między klasami K przy użyciu ważonej sumy zmiennych predykcyjnych. Oblicza prawdopodobieństwo wystąpienia zdarzenia i może służyć do wykrywania raka.
Biopython zapewnia moduł Bio.LogisticRegression do przewidywania zmiennych w oparciu o algorytm regresji logistycznej. Obecnie Biopython implementuje algorytm regresji logistycznej tylko dla dwóch klas (K = 2).
k-Najbliżsi sąsiedzi
k-Nearest neighbors to również nadzorowany algorytm uczenia maszynowego. Działa poprzez kategoryzowanie danych na podstawie najbliższych sąsiadów. Biopython dostarcza moduł Bio.KNN do przewidywania zmiennych w oparciu o algorytm k-najbliższych sąsiadów.
Naiwny Bayes
Naiwne klasyfikatory Bayesa to zbiór algorytmów klasyfikacyjnych opartych na twierdzeniu Bayesa. Nie jest to pojedynczy algorytm, ale rodzina algorytmów, w których wszystkie mają wspólną zasadę, tj. Każda para klasyfikowanych cech jest od siebie niezależna. Biopython dostarcza moduł Bio.NaiveBayes do pracy z algorytmem Naive Bayes.
Model Markowa
Model Markowa to system matematyczny zdefiniowany jako zbiór zmiennych losowych, który doświadcza przejścia z jednego stanu do drugiego zgodnie z określonymi regułami probabilistycznymi. Biopython zapewniaBio.MarkovModel and Bio.HMM.MarkovModel modules to work with Markov models.
Biopython ma rozbudowany skrypt testowy do testowania oprogramowania w różnych warunkach, aby upewnić się, że oprogramowanie jest wolne od błędów. Aby uruchomić skrypt testowy, pobierz kod źródłowy Biopythona, a następnie uruchom poniższe polecenie -
python run_tests.pySpowoduje to uruchomienie wszystkich skryptów testowych i da następujące dane wyjściowe -
Python version: 2.7.12 (v2.7.12:d33e0cf91556, Jun 26 2016, 12:10:39)
[GCC 4.2.1 (Apple Inc. build 5666) (dot 3)]
Operating system: posix darwin
test_Ace ... ok
test_Affy ... ok
test_AlignIO ... ok
test_AlignIO_ClustalIO ... ok
test_AlignIO_EmbossIO ... ok
test_AlignIO_FastaIO ... ok
test_AlignIO_MauveIO ... ok
test_AlignIO_PhylipIO ... ok
test_AlignIO_convert ... ok
...........................................
...........................................Możemy również uruchomić indywidualny skrypt testowy, jak określono poniżej -
python test_AlignIO.pyWniosek
Jak się dowiedzieliśmy, Biopython jest jednym z ważnych programów w dziedzinie bioinformatyki. Napisany w Pythonie (łatwy do nauczenia i pisania), zapewnia rozbudowaną funkcjonalność do obsługi wszelkich obliczeń i operacji z zakresu bioinformatyki. Zapewnia również łatwy i elastyczny interfejs dla prawie każdego popularnego oprogramowania bioinformatycznego, aby wykorzystać jego funkcjonalność.