ヒトゲノムプロジェクトが2003年に最初のヒトゲノムを完成させたと発表したとき、それは重大な成果でした—初めて、人間の生命のDNA青写真が解き放たれました。しかし、それは落とし穴がありました—彼らは実際にゲノム内のすべての遺伝情報をまとめることができませんでした。ギャップがありました:埋められていない、しばしば繰り返しの領域は、あまりにも混乱してつなぎ合わせることができませんでした。
これらの反復配列を処理できる技術の進歩により、科学者はついに2021年5月にこれらのギャップを埋め、最初のエンドツーエンドのヒトゲノムが2022年3月31日に正式に公開されました。
私はゲノム生物学者であり、反復DNA配列と、それらが進化の歴史を通じてどのようにゲノムを形成するかを研究しています。私は、ゲノムから欠落している反復配列の特徴を明らかにするのを手伝ったチームの一員でした。そして今、真に完全なヒトゲノムにより、これらの発見された反復領域がついに初めて完全に探索されています。
行方不明のパズルのピース
ドイツの植物学者ハンス・ヴィンクラーは、1920年に「ゲノム」という単語を作り出し、「遺伝子」という単語と「完全なセット」を意味する接尾辞「-ome」を組み合わせて、各セルに含まれる完全なDNA配列を説明しました。研究者たちは、生物を構成する遺伝物質を指すために、1世紀後もこの言葉を使用しています。
ゲノムがどのように見えるかを説明する1つの方法は、それを参考書と比較することです。この例えでは、ゲノムは生命のDNA命令を含むアンソロジーです。これは、染色体(章)にパッケージ化された膨大な数のヌクレオチド(文字)で構成されています。各染色体には、生物が機能することを可能にする特定のタンパク質をコードするDNAの領域である遺伝子(段落)が含まれています。
すべての生物にはゲノムがありますが、そのゲノムのサイズは種によって異なります。象は、食べる草や腸内のバクテリアと同じ形の遺伝情報を使用します。しかし、2つのゲノムがまったく同じように見えることはありません。昆虫に生息するバクテリアNasuiadeltocephalinicolaのゲノムのように、 112,000ヌクレオチドにわたってわずか137個の遺伝子を持つ短いものもあります。顕花植物キヌガサソウの1490億ヌクレオチドのように、非常に長いため、中に含まれている遺伝子の数を把握するのが難しいものもあります。
しかし、タンパク質をコードするDNAのストレッチとして、伝統的に理解されてきた遺伝子は、生物のゲノムのほんの一部にすぎません。実際、それらはヒトDNAの2パーセント未満を構成します。
ヒトゲノムには、およそ30億のヌクレオチドと、20,000をわずかに下回るタンパク質をコードする遺伝子が含まれています。これは、ゲノムの全長の推定1パーセントです。残りの99%は、タンパク質を生成しない非コードDNA配列です。一部は、他の遺伝子がどのように機能するかを制御するための交換機として機能する規制コンポーネントです。その他は、偽遺伝子、または機能する能力を失ったゲノム遺物です。
そして、ヒトゲノムの半分以上が反復的であり、ほぼ同一の配列の複数のコピーがあります。
反復DNAとは何ですか?
反復DNAの最も単純な形式は、衛星と呼ばれるタンデムで何度も繰り返されるDNAのブロックです。特定のゲノムが持つサテライトDNAの量は人によって異なりますが、テロメアと呼ばれる領域の染色体の端に向かってクラスター化することがよくあります。これらの領域は、DNA複製中に染色体が分解するのを防ぎます。それらは染色体のセントロメアにも見られます。これは、細胞が分裂したときに遺伝情報を無傷に保つのに役立つ領域です。
研究者はまだサテライトDNAのすべての機能の明確な理解を欠いています。しかし、サテライトDNAは各人に固有のパターンを形成するため、法医学の生物学者や遺伝学者はこのゲノムの「指紋」を使用して犯罪現場のサンプルを照合し、祖先を追跡します。50以上の遺伝性疾患は、ハンチントン病を含むサテライトDNAの変異に関連しています。
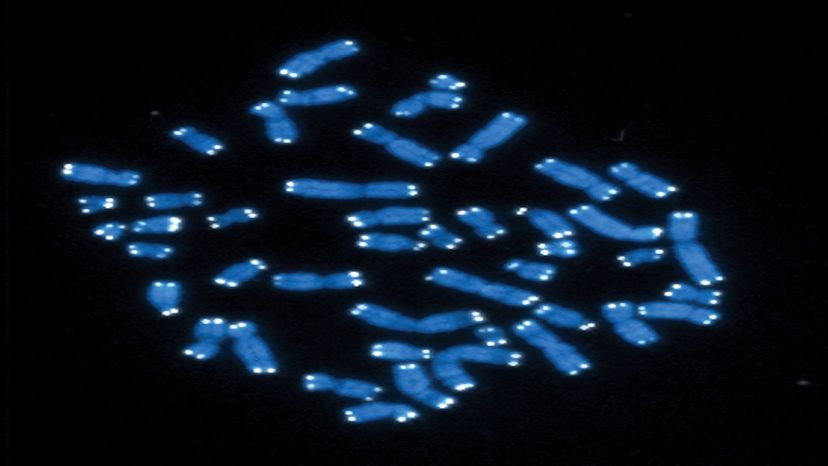
別の豊富なタイプの反復DNAは、トランスポゾン、またはゲノム内を移動できる配列です。
一部の科学者は、結果に関係なく、ゲノムのどこにでも自分自身を挿入できるため、利己的DNAと表現しています。ヒトゲノムが進化するにつれて、多くの転移因子は、有害な中断を回避するために動く能力を抑制する突然変異を収集しました。しかし、まだ動き回ることができる人もいます。たとえば、転移因子の挿入は、遺伝性出血障害である血友病Aの多くの症例に関連しています。
しかし、転移因子は破壊的なだけではありません。それらは、他のDNA配列の発現を制御するのに役立つ調節機能を持つことができます。それらがセントロメアに集中している場合、それらは細胞の生存に不可欠な遺伝子の完全性を維持するのにも役立つ可能性があります。
それらはまた進化に貢献することができます。研究者たちは最近、発生に重要な遺伝子への転移因子の挿入が、人間を含む一部の霊長類がもはや尾を持たない理由である可能性があることを発見しました。転移因子による染色体再配列は、東南アジアのギボンやオーストラリアのワラビーのような新種の起源にさえ関連しています。
ゲノムパズルを完成させる
最近まで、これらの複雑な領域の多くは月の裏側と比較することができました。存在することが知られていますが、見えません。
1990年にヒトゲノムプロジェクトが最初に開始されたとき、技術的な制限により、ゲノム内の反復領域を完全に明らかにすることは不可能でした。利用可能なシーケンシングテクノロジーは、一度に約500ヌクレオチドしか読み取ることができず、完全なシーケンスを再作成するには、これらの短いフラグメントを互いにオーバーラップさせる必要がありました。研究者は、これらの重複するセグメントを使用して、シーケンス内の次のヌクレオチドを識別し、一度に1フラグメントずつゲノムアセンブリを段階的に拡張しました。
これらの繰り返しのギャップ領域は、どんよりした空の1,000ピースのパズルを組み立てるようなものでした。すべてのピースが同じように見える場合、ある雲がどこから始まり、別の雲がどこで終わるかをどうやって知るのでしょうか。多くのスポットでほぼ同一のオーバーラップストレッチがあるため、ゲノムを断片的に完全にシーケンスすることは不可能になりました。ヒトゲノムの最初の反復では、何百万ものヌクレオチドが隠されたままでした。
それ以来、シーケンスパッチはヒトゲノムのギャップを少しずつ埋めていきました。そして2021年、テロメアからテロメア(T2T)コンソーシアムは、ヒトゲノムの組み立てを端から端まで完了するために活動している科学者の国際コンソーシアムであり、残りのすべてのギャップがついに埋められたと発表しました。
これは、数千ヌクレオチドの長さのより長い配列を読み取ることができる改良された配列決定技術によって可能になりました。より大きな画像内に反復配列を配置するためのより多くの情報により、ゲノム内のそれらの適切な位置を特定することがより簡単になりました。1,000ピースのパズルを100ピースのパズルに単純化するように、長い読み取りシーケンスにより、初めて大きな反復領域を組み立てることが可能になりました。
長い間読まれてきたDNAシーケンシング技術の力が増すにつれ、遺伝学者はゲノミクスの新時代を探求し、集団や種にまたがる複雑な反復配列を初めて解き明かす立場にあります。そして、完全でギャップのないヒトゲノムは、研究者が遺伝的構造と変異、種の進化と人間の健康を形作る反復領域を調査するための貴重なリソースを提供します。
しかし、1つの完全なゲノムがすべてを捉えているわけではありません。努力は、地球上の人口と生命を完全に表す多様なゲノム参照を作成し続けています。より完全な「テロメアからテロメアへ」のゲノム参照により、科学者はDNAの反復的な暗黒物質をより明確に理解できるようになります。
GabrielleHartleyは博士号を取得しています。コネチカット大学で分子生物学および細胞生物学の候補者。彼女は国立科学財団から資金提供を受けています。
この記事は、クリエイティブコモンズライセンスの下で会話から再発行されています。元の記事はここにあります。